UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 20-F
(Mark One)
REGISTRATION STATEMENT PURSUANT TO SECTION 12(b) OR (g) OF THE SECURITIES EXCHANGE ACT OF 1934 |
OR
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31 , 2025
OR
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from _____________________ to _____________________
OR
SHELL COMPANY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Date of event requiring this shell company report: _____________________
Commission file number 001-42128
(Exact name of Registrant as specified in its charter)
N/A
(Translation of Registrant's name into English)
(Jurisdiction of incorporation or organization)
(Address of principal executive offices)
Tel: +61 3 9093 3855
(Name, Telephone, E-mail and/or Facsimile number and Address of Company Contact Person)
Securities registered or to be registered pursuant to Section 12(b) of the Act:
Title of each class | Trading Symbol | Name of each exchange on which registered | ||
representing one ordinary share, no par value Ordinary shares, no par value* |
•Listed not for trading, but only in connection with the listing of the American Depositary Shares, pursuant to the
requirements of the Securities & Exchange Commission
Securities registered or to be registered pursuant to Section 12(g) of the Act: None.
Securities for which there is a reporting obligation pursuant to Section 15(d) of the Act: None.
Indicate the number of outstanding shares of each of the issuer’s classes of capital or common stock as of the close of
the period covered by the annual report: The number of ordinary shares outstanding as of December 31, 2025 was
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
If this report is an annual or transition report, indicate by check mark if the registrant is not required to file reports
pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934. Yes o No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the
Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was
required to file such reports), and (2) has been subject to such filing requirements for the past 90 days: Yes x No o
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be
submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such
shorter period that the registrant was required to submit such files): Yes x No o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or
an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” and “emerging growth
company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer | x | Accelerated filer | o | Non-accelerated filer | o | Emerging growth company |
If an emerging growth company that prepares its financial statements in accordance with U.S. GAAP, indicate by check
mark if the registrant has elected not to use the extended transition period for complying with any new or revised
financial accounting standards† provided pursuant to Section 13(a) of the Exchange Act. ☐
† The term “new or revised financial accounting standard” refers to any update issued by the Financial Accounting
Standards Board to its Accounting Standards Codification after April 5, 2012.
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of
the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15
U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. x
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements
of the registrant included in the filing reflect the correction of an error to previously issued financial statements. o
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of
incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period
pursuant to §240.10D-1(b). o
Indicate by check mark which basis of accounting the registrant has used to prepare the financial statements included in
this filing:
U.S. GAAP o
Other o
If “Other” has been checked in response to the previous question, indicate by check mark which financial statement item
the registrant has elected to follow. Item 17 o Item 18 o
If this is an annual report, indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of
the Exchange Act). Yes o No x
TABLE OF CONTENTS
1
INTRODUCTION
Telix Pharmaceuticals Limited was incorporated under the laws of Australia in January 2017. Our ordinary shares have
been listed on the Australian Securities Exchange ("ASX") since November 2017, and since November 2024, have been
listed on the Nasdaq Global Select Market in the form of American depositary shares ("ADSs"), with each ADS
representing one of our ordinary shares. JP Morgan Chase Bank, N.A., acts as depositary for the ADSs. Throughout this
annual report, all references to “ADRs” mean the American depositary receipts that evidence the ADSs.
As of January 1, 2025, our reporting currency is the United States Dollar ("US$"). All amounts presented in this Annual
Report on Form 20-F ("Annual Report") are presented in US$ unless otherwise indicated. Prior to January 1, 2025, our
presentation currency was the Australian Dollar. The change in presentation currency has been applied retrospectively to
all comparative periods presented See Note 2.3.1 to our audited consolidated financial statements included elsewhere in
this Annual Report.
The consolidated financial statements and related notes included elsewhere in this Annual Report have been prepared in
accordance with International Financial Reporting Standards as issued by the International Accounting Standards Board,
(“IFRS Accounting Standards”), which differ in certain significant respects from generally accepted accounting principles
in the United States ("U.S. GAAP").
We also maintain a website at www.telixpharma.com. The information contained on our website or available through our
website is not incorporated by reference into and should not be considered a part of this Annual Report, and any
references to our website throughout this Annual Report are inactive textual references only.
Unless otherwise stated or the context indicates otherwise, all references herein to “Telix,” “Telix Pharmaceuticals,” "the
Company,” "the Group," “our company,” “we,” “us,” “our” and similar references refer to Telix Pharmaceuticals Limited
and its consolidated subsidiaries, taken as a whole.
Australian Disclosure Requirements
Our ordinary shares are primarily quoted on the ASX in addition to our listing of our ADSs on the Nasdaq Global Select
Market. As part of our ASX listing, we are required to comply with various disclosure requirements as set out under the
Australian Corporations Act 2001 (Cth) and the ASX Listing Rules. Information furnished under the sub-heading
“Australian Disclosure Requirements” is intended to comply with ASX listing and Australian Corporations Act 2001 (Cth)
disclosure requirements and is not intended to fulfill information required by this Annual Report.
INDUSTRY AND MARKET DATA
This Annual Report contains estimates and information concerning our industry and our business, including estimated
market size and projected growth rates of the markets for our product candidates. Unless otherwise expressly stated,
we obtained this industry, business, market, medical and other information from reports, research surveys, studies and
similar data prepared by third parties, industry, medical and general publications, government data and similar sources.
This information involves a number of assumptions and is based on limited available information. Although we are
responsible for all of the disclosure contained in this Annual Report and we believe the third-party market position,
market opportunity and market size data included in this Annual Report are reliable, we have not independently verified
the accuracy or completeness of this third-party data. In addition, projections, assumptions and estimates of our future
performance and the future performance of the industry in which we operate are necessarily subject to a high degree of
uncertainty and risk due to a variety of factors, including those described in “Item 3. Key Information — D. Risk Factors.”
These and other factors could cause results to differ materially from those expressed in these publications and reports.
TRADEMARKS AND SERVICE MARKS
Telix Pharmaceuticals, the Telix logo and other trademarks or service marks of Telix appearing in this Annual Report are
the property of Telix or its subsidiaries. Solely for convenience, the trademarks, service marks and trade names referred
to in this Annual Report are listed without the ® and ™ symbols, but such references should not be construed as any
indicator that their respective owners will not assert, to the fullest extent under applicable law, their right thereto. All
other trademarks, trade names and service marks appearing in this Annual Report are the property of their respective
owners. Any use or display of any third-party trademarks, trade names or service marks is not intended to imply a
relationship with, endorsement or sponsorship of or by the respective owners.
2
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report contains forward-looking statements about us and our industry that involve substantial risks and
uncertainties. All statements other than statements of historical facts contained in this Annual Report, including
statements regarding our future results of operations, financial condition, business strategy, prospective products,
product approvals, research and development costs, future revenue and plans and objectives of management for future
operations, are forward-looking statements. In some cases, you can identify forward-looking statements because they
contain words such as “anticipate,” “believe,” “contemplate,” “continue,” “could,” “estimate,” “expect,” “intend,” “may,”
“plan,” “potential,” “predict,” “project,” “should,” “target,” “will,” or “would,” or the negative of these words or other similar
terms or expressions. We have based these forward-looking statements largely on our current expectations and
projections about future events and trends that we believe may affect our financial condition, results of operations,
business strategy and financial needs. These forward-looking statements are subject to a number of known and
unknown risks, uncertainties, other factors and assumptions, including the risks described in “Item 3. Key Information —
D. Risk Factors” and elsewhere in this Annual Report, regarding, among other things:
•the ongoing commercialization of our Commercial Products and our preparation for the commercialization of our
product candidates, if they are approved;
•the timing and review of submissions for regulatory approval of our product candidates, including review of our
resubmissions for Pixclara® (TLX101-Px) and Zircaix® (TLX250-Px), and our ability to obtain and maintain such
regulatory approvals;
•the initiation, timing, progress and results of our ongoing and planned clinical trials, including the timing of dosing of
patients, enrollment and completion of these trials, including multi-national trials, and the anticipated results from
these trials;
•our sales, marketing and distribution capabilities and strategies, including for the commercialization and
manufacturing of our Commercial Products and any future products;
•our ability to obtain and maintain an adequate supply at reasonable costs of raw materials we may incorporate into
our products and product candidates;
•our ability to address the fulfillment and logistical challenges posed by the time-limited stabilization of our products
and product candidates;
•our commercialization, marketing and manufacturing capabilities and strategy, including the timing and costs of
expanding our manufacturing capabilities;
•the rate and degree of market acceptance and clinical utility of our products and product candidates, if they are
approved;
•the pricing and reimbursement of our products and product candidates, if and after they have been approved;
•estimates of our expenses, future revenues and capital requirements;
•our financial performance;
•developments relating to our competitors and industry;
•the success of our collaborations and partnerships with third parties;
•our ability to maintain, expand, protect and enforce our regulatory exclusivity and intellectual property ("IP")
portfolio;
•our expectations regarding our ability to obtain and maintain regulatory exclusivity and intellectual property
protection for our products and product candidates;
•our ability to successfully integrate the businesses that we have acquired or may acquire in the future;
•our estimates regarding expenses, future revenues, capital requirements and needs for additional financing;
•changes to law, policy and regulation in the U.S., Australia and other jurisdictions;
•disruptions caused by the current U.S. presidential administration or as a result of legislative or judicial action or lack
thereof, including at the FDA and other government agencies;
•our ability to remain compliant with the respective listing rules and standards of the ASX, the Singapore Exchange
Securities Trading Limited ("SGX"), and the Nasdaq Global Select Market ("Nasdaq");
3
•our ability to attract and retain key scientific or management personnel;
•the success of competing therapies that are or may become available;
•the volatility of currency exchange rates;
•the impact of and changes in governmental regulations or the enforcement thereof, tax laws and rates, accounting
guidance and similar matters in regions in which we operate or will operate in the future;
•any changes in laws, rules or regulations, including the imposition of tariffs or other trade restrictions, affecting our
ability to manufacture, test, develop, or commercialize our Commercial Products and product candidates;
•changes to staffing, process, or policy at government agencies, including, but not limited to, the FDA; and
•changes in U.S. and international trade policies, including the imposition of tariffs on raw materials and finished
products; and
•other risks and uncertainties, including those listed under “Item 3. Key Information — D. Risk Factors.”
These risks are not exhaustive. Other sections of this Annual Report may include additional factors that could harm our
business and financial performance. New risk factors may emerge from time to time and it is not possible for our
management to predict all risk factors, nor can we assess the impact of all factors on our business or the extent to which
any factor, or combination of factors, may cause actual results to differ materially from those contained in, or implied by,
any forward-looking statements.
You should not rely on forward-looking statements as predictions of future events. We have based the forward-looking
statements contained in this Annual Report primarily on our current expectations and projections about future events and
trends that we believe may affect our business, financial condition and operating results. We undertake no obligation to
update any forward-looking statements made in this Annual Report to reflect events or circumstances after the date of
this Annual Report or to reflect new information or the occurrence of unanticipated events, except as required by law.
We may not actually achieve the plans, intentions or expectations disclosed in our forward-looking statements, and you
should not place undue reliance on our forward-looking statements. Our forward-looking statements do not reflect the
potential impact of any future acquisitions, mergers, dispositions, joint ventures or investments.
In addition, statements that “we believe” and similar statements reflect our beliefs and opinions on the relevant subject.
These statements are based on information available to us as of the date of this Annual Report. While we believe that
information provides a reasonable basis for these statements, that information may be limited or incomplete. Our
statements should not be read to indicate that we have conducted an exhaustive inquiry into, or review of, all relevant
information. These statements are inherently uncertain, and investors are cautioned not to unduly rely on these
statements.
You should read this Annual Report and the documents that we reference and have filed as exhibits to the Annual Report
with the understanding that our actual future results, performance and achievements may be different from what we
expect. We qualify all of our forward-looking statements by these cautionary statements. In addition, with respect to all
of our forward-looking statements, we claim the protection of the safe harbor for forward-looking statements contained
in the Private Securities Litigation Reform Act of 1995.
4
PART I
ITEM 1.IDENTITY OF DIRECTORS, SENIOR MANAGEMENT AND ADVISERS
A.Directors and Senior Management
Not applicable.
B.Advisers
Not applicable.
C.Auditors
Not applicable.
ITEM 2.OFFER STATISTICS AND EXPECTED TIMETABLE
A.Offer Statistics
Not applicable.
B.Method and Expected Timetable
Not applicable.
ITEM 3.KEY INFORMATION
A.Reserved
B.Capitalization and Indebtedness
Not applicable.
C.Reasons for the Offer and Use of Proceeds
Not applicable.
D.Risk Factors
Investing in our securities involves a high degree of risk. You should consider and read carefully all of the risks and
uncertainties described below, as well as other information included in this Annual Report, including our consolidated
financial statements and related notes included elsewhere in this Annual Report, before making an investment decision. If
any of the following risks actually occur, it could harm our business, prospects, results of operations and financial
condition. In such event, the trading price of our ordinary shares and the ADSs could decline, and you might lose all or
part of your investment.
Risk Factors Summary
•We have a history of significant net losses, our operating expenses may increase in the future, and we may not be
able to maintain profitability in future periods.
•We may need to raise capital to achieve our business objectives if we are unable to fund our operations with our
cash flows from the sale of our products. If we are unable to raise capital when needed or on acceptable terms, we
would be forced to delay, reduce or eliminate our research and development programs and/or commercialization
efforts.
•We may not be able to effectively integrate the businesses that we have acquired and/or may acquire in the future.
•Our business is substantially dependent on the commercial success of our Commercial Products and our product
candidates, if approved. If we are unable to successfully commercialize our Commercial Products as currently
5
approved or to successfully commercialize our product candidates, if approved, our business, financial condition
and results of operations will be materially harmed.
•Clinical development is a lengthy and expensive process, with uncertain timelines and outcomes. If clinical trials of
our product candidates fail to demonstrate safety and efficacy to the satisfaction of regulatory authorities or do not
otherwise produce positive results, we may incur additional costs or experience delays in completing, or ultimately
be unable to complete, the development and commercialization of such product candidates, if approved.
•The results of previous clinical trials may not be predictive of future trial results, and preliminary, interim or top-line
data may be subject to change or qualification based on the complete analyses of data and, therefore, may not be
predictive of the final results of a trial.
•We may find it difficult to enroll patients in our clinical trials. If we encounter difficulties enrolling subjects in our
clinical trials, our clinical development activities could be delayed or otherwise adversely affected.
•Due to their radioactive nature, our Commercial Products and our product candidates have time-limited stability,
and as a result, we may encounter difficulties with fulfillment and logistics.
•We face substantial competition, which may result in others discovering, developing, or commercializing products
before or more successfully than we do.
•The commercial success of our Commercial Products and our product candidates, if approved, will depend upon
public perception of radiopharmaceuticals and the degree of their market acceptance by physicians, patients,
healthcare payors and others in the medical community.
•We may be unable to generate and/or obtain a sufficient supply of radioisotopes to support clinical development or
manufacturing at commercial scale.
•Even if we are able to effectively commercialize our Commercial Products or any product candidates for which we
obtain approval, the products may not receive coverage or may become subject to unfavorable pricing regulations,
third-party reimbursement practices or healthcare reform initiatives, all of which would harm our business.
•We depend on collaborations with third parties for certain aspects of the development, marketing and/or
commercialization of our Commercial Products and our product candidates. If those collaborations are not
successful, or if we are not able to maintain our existing collaborations or establish additional collaborations, we
may have to alter our development and commercialization plans and may not be able to capitalize on the market
potential of our Commercial Products or our product candidates, if approved.
•If we are unable to obtain and/or maintain commercially valuable regulatory exclusivity and intellectual property, or
to protect our patents, trademarks, know-how and trade secrets, our ability to successfully commercialize our
Commercial Products and product candidates, if approved, would be adversely impacted.
•We may experience sustainability risks, including: physical climate-related risks such as extreme weather events
that could disrupt internal operations, suppliers, and logistics, and increase operational costs; resource and energy
risks such as increasing energy prices, regulatory actions and increased resource efficiency standards that may
increase operating expenses and capital expenditure for facility upgrades; and, non-compliance with increased
social and governance regulations that could lead to fines, reputational damage, and increased compliance costs.
•An active and liquid market for our securities may not continue to be developed or sustained, which could harm the
market price of the ADSs.
•As a foreign private issuer, we are permitted to follow certain home country corporate governance practices in lieu
of certain Nasdaq requirements applicable to domestic issuers.
•We have identified a material weakness in our internal control over financial reporting. If we are unable to remediate
this material weaknesses, or if we identify additional material weaknesses in the future or otherwise fail to maintain
an effective system of internal controls, we may not be able to accurately or timely report our financial condition or
results of operations, which may adversely affect investor confidence in us and, as a result, the value of our ADSs.
Risks Related to Our Financial Position and Capital Requirements
We have a history of significant net losses, our operating expenses may increase in the future, and we may not be
able to maintain profitability in future periods.
Until 2023, we incurred significant operating losses. Our loss after tax was $7.1 million for the year ended December 31,
2025. Our net operating cash flow was $17.3 million (outflow) for the year ended December 31, 2025. As of
December 31, 2025, we had an accumulated deficit of $154.5 million. Although we launched Illuccix in April 2022 and
Gozellix in June 2025 and have recognized profits in recent periods, we cannot be certain that we will sustain profitability
or positive cash flows from operations in future periods.
6
We have invested most of our resources in developing our technology and product candidates, building our intellectual
property portfolio, developing our supply chain, conducting business planning, raising capital and providing general and
administrative support for these operations. We continue to incur significant research and development ("R&D"), and
other expenses related to ongoing operations and may incur losses in the future.
Investment in biotechnology product development, as well as medical device development, is highly speculative because
it entails substantial upfront capital expenditures and significant risk that any potential product candidate will be unable
to demonstrate effectiveness or an acceptable safety profile, gain regulatory approval, gain competitive pricing or
reimbursement and become commercially viable.
Illuccix has been approved by the FDA, the Australian Therapeutic Goods Administration ("TGA"), by Health Canada, by
the Brazilian Health Regulatory Agency ("ANVISA"), by the United Kingdom Medicines and Healthcare Products
Regulatory Agency ("MHRA") and by health regulators in 19 European Economic Area member states. Gozellix was
approved by the FDA in March 2025. Throughout this Annual Report, “Commercial Products” is used to refer to Illuccix
and Gozellix collectively.
We have historically financed our operations principally through product sales, private and institutional placements of our
ordinary shares, proceeds from our initial public offering of ordinary shares on the ASX, proceeds from our issuance of
convertible bonds, loan agreements with financial institutions and cash generated from our business development
activities. Substantially all of our operating losses in previous periods have resulted from costs incurred in connection
with our research and development programs, the pursuit of regulatory approvals within and outside of the U.S., and the
commercialization of our Commercial Products. We expect to continue to incur significant expenses as we continue to
commercialize our Commercial Products in the jurisdictions in which we have received marketing authorization, and other
jurisdictions, if approved, and engage in activities to prepare for the potential approval and commercialization of our
product candidates. The profits or losses we incur may fluctuate significantly from quarter to quarter and year to year.
While we began to generate revenue from the sales of Illuccix in April 2022 and from Gozellix in June 2025, there can be
no assurance as to the amount or timing of future product or license and other revenues, and we may not be able to
maintain profitability in future periods. Our ability to remain profitable depends significantly on our success in many
areas, including:
•effectively commercializing Illuccix, Gozellix or any future products either on our own or with a collaborator,
including by maintaining a full commercial organization required to market, sell and distribute our products, and
achieving an adequate level of market acceptance;
•the impact of current or future competing products on product sales of Illuccix, Gozellix or any future products;
•obtaining sufficient pricing, coverage and reimbursement, under U.S. federal healthcare programs, such as
Medicare and Medicaid, and from private payors, for Illuccix, Gozellix and any future products from private and
government payors and the impact of any pricing changes;
•initiating and successfully completing clinical trials required to file for, obtain and maintain regulatory approval for
our product candidates;
•obtaining and maintaining regulatory approvals, and the timing of such approvals;
•manufacturing at commercial scale;
•establishing and managing any collaborations for the development, marketing and/or commercialization of our
products and product candidates, if approved, including the level of success of any such collaborators’ efforts and
the timing and amount of any milestone or royalty payments we may receive; and
•obtaining, maintaining and protecting our intellectual property rights.
We anticipate that our operating expenses will continue to be significant and increase as we continue to:
•commercialize Illuccix and Gozellix in the U.S., and Illuccix in Australia, New Zealand, Brazil, Canada, Europe and
other jurisdictions following regulatory approval, including maintaining our commercial infrastructure;
•obtain and/or maintain regulatory approval for Illuccix, Gozellix and our product candidates, including completing
any required post-marketing requirements to the satisfaction of the FDA or other regulatory agencies;
•expand our research and development programs, identify additional product candidates and initiate and conduct
clinical trials, including clinical trials required by the FDA or other regulatory agencies in addition to those that have
been or are currently expected to be conducted;
•maintain, expand and protect our intellectual property portfolio;
•manufacture Illuccix, Gozellix and our product candidates;
7
•add clinical, scientific, operational, financial and management information systems and personnel, including
personnel to support our product development and potential future radiopharmaceutical commercialization efforts;
•operate as a publicly listed company in the U.S. and Australia; and
•acquire or in-license other products, product candidates or technologies.
Because of the numerous risks and uncertainties associated with pharmaceutical product development and
commercialization, we are unable to accurately predict the timing or amount of our revenue and expenses or if we will be
able to maintain profitability. We cannot be certain that our revenue from sales of Illuccix and Gozellix, in the currently
approved indications, will be sufficient for us to remain profitable in future periods. We may not generate revenues that
are significant or large enough to sustain or increase profitability on an annual basis. Our failure to remain profitable
would decrease the value of our company and could impair our ability to raise capital, maintain our research and
development and commercialization efforts, expand our business and/or continue our operations. This could result in a
material adverse effect on the value of our company and could cause our shareholders and ADS holders to lose all or part
of their investment.
We may need to raise additional capital to achieve our business objectives if we are unable to fund our operations
with our cash flows from the sale of our products. If we are unable to raise capital when needed or on acceptable
terms, we would be forced to delay, reduce or eliminate our research and development programs and/or
commercialization efforts.
Discovering, developing and commercializing products involve time-consuming, expensive and uncertain processes that
take years to complete. We have used substantial funds to develop Illuccix and Gozellix and expect our operating
expenses to continue to increase as we continue to commercialize Illuccix, Gozellix or any future approved products,
conduct further research and development of our product candidates, seek approval and prepare for commercialization
of TLX250-Px and TLX101-Px and continue to conduct clinical trials for our other product candidates.
Furthermore, we will continue to incur additional costs associated with operating as a public company, hiring additional
personnel and expanding our geographical reach. Although currently Illuccix is commercially available in multiple
jurisdictions worldwide and Gozellix is commercially available in the U.S., we cannot be certain that our revenue from
product sales of Illuccix and Gozellix will be sufficient for us to remain profitable on an annual basis. Accordingly, we may
need to continue to rely on additional financing to achieve our business objectives.
As of December 31, 2025, we had $141.9 million in cash and cash equivalents. The amount and timing of our future
capital requirements will depend on many factors, including, but not limited to:
•the scope, progress, results, timing and costs of our current and planned development efforts and regulatory review
of our product candidates;
•the amount and timing of revenues from sales of Illuccix, Gozellix or any product candidate for which we receive
regulatory approval;
•the cost of, and our ability to expand and maintain, the commercial infrastructure required to support the
commercialization of Illuccix, Gozellix and any other product for which we receive regulatory approval, including
medical affairs, manufacturing, marketing and distribution functions;
•our ability to establish and maintain collaboration, partnership, licensing, marketing, distribution or other
arrangements on favorable terms and the level and timing of success of these arrangements;
•the extent to which we acquire or in-license other products, product candidates and technologies; and
•the costs and timing of preparing, filing and prosecuting patent applications, maintaining and enforcing our
intellectual property rights and defending intellectual property-related claims.
In addition, the terms of any financing may adversely affect the holdings or the rights of our shareholders and ADS
holders. If we raise funds by issuing equity securities, dilution to our existing shareholders and ADS holders will result,
and this may also have an impact on the market price of our ordinary shares and ADSs. In addition, as a condition to
providing additional funding to us, future investors may demand, and may be granted, rights superior to those of existing
shareholders. Moreover, any debt financing, if available, may involve restrictive covenants that could limit our flexibility in
conducting future business activities and, in the event of insolvency, would be paid before holders of equity securities
received any distribution of corporate assets. Our ability to satisfy and meet any future debt service obligations will
depend upon our future performance, which will be subject to financial, business and other factors affecting our
operations, many of which are beyond our control.
Even if we believe we have sufficient funds for our current or future operating plans, we may seek additional capital due
to favorable market conditions or strategic considerations. Any future fundraising efforts could divert our management’s
attention away from their day-to-day activities. Further, adequate additional financing may not be available to us on
acceptable terms, or at all. In addition, raising funds in the current economic environment may present additional
8
challenges. For example, any sustained disruption in the capital markets from adverse macroeconomic conditions, such
as the disruption and uncertainty caused by rising inflation, increasing interest rates and slower economic growth or
recession, could negatively impact our ability to raise capital and we cannot predict the extent or duration of such
macro-economic disruptions. If adequate funds are not available to us on a timely basis or on attractive terms, we may
be required to delay, reduce or eliminate our research and development programs or any current or future
commercialization efforts for one or more of our products or product candidates, any of which could have a material
adverse effect on our business, operating results and prospects.
Our operating results may fluctuate significantly or may fall below the expectations of investors or securities
analysts, each of which may cause the trading price of our ordinary shares and the ADSs to fluctuate or decline.
We expect our operating results to be subject to fluctuations. Our profit or loss and other operating results will be
affected by numerous factors, including:
•timing and variations in the level of expense related to the current or future development of our programs;
•timing and status of enrollment for our clinical trials;
•results of clinical trials, or the addition or termination of clinical trials or funding support by us or potential future
partners;
•timing of any milestone payments or other payment obligations to be paid by us pursuant to existing supply
agreements, licenses or collaborations;
•timing of any milestone payments or other payments to be received by us pursuant to our license agreement;
•our execution of any collaboration, licensing or similar arrangements, and the timing of payments we may make or
receive under potential future arrangements or the termination or modification of any such potential future
arrangements;
•any intellectual property infringement, misappropriation or violation lawsuit or opposition, or other post-grant
proceeding or cancellation proceeding in which we may become involved;
•additions and departures of key personnel;
•strategic decisions, such as acquisitions, divestitures, spin-offs, joint ventures, strategic investments or changes in
business strategy;
•if any product candidate we may develop receives regulatory approval, the timing and terms of such approval and
market acceptance and demand for such product candidate;
•the timing and cost to establish a sales, marketing and distribution infrastructure to commercialize any products or
product candidates for which we may obtain regulatory approval and intend to commercialize on our own or jointly
with current or future collaborators;
•regulatory developments affecting our Commercial Products or any other of our product candidates or those of our
competitors, including disruptions at FDA or other government agencies; and
•changes in general market and economic conditions, including as a result tariffs, trade restrictions, conflicts or other
geopolitical risks that may arise.
If our operating results fall below the expectations of investors or securities analysts, the price of our ordinary shares and
ADSs could decline substantially. Furthermore, any fluctuations in our operating results may, in turn, cause the price of
our ordinary shares and ADSs to fluctuate substantially. We believe that comparisons of our financial results are not
necessarily meaningful and should not be relied upon as an indication of our future performance.
Our Loan Agreements with BNP Paribas and IMBC Group contain various covenants and other provisions, which, if
violated, could result in the acceleration of payments due under such agreement, as well as affect the buildout of our
Brussels South manufacturing facility.
In March 2022, one of our subsidiaries, Telix Pharmaceuticals (Belgium) SPRL (now "Telix Pharmaceuticals (Belgium)
SRL"), entered into Loan Agreements ("the Loan Agreements") with BNP Paribas and IMBC Group. The borrowings under
these Loan Agreements were used to fund in part the construction of our Brussels South manufacturing facility. Pursuant
to the Loan Agreements, Telix Pharmaceuticals (Belgium) SRL is required to comply with various covenants relating to
the conduct of its business. The Loan Agreements also include customary events of default upon the occurrence of
enumerated events, including non-payment of required repayments, failure to perform certain covenants and the
occurrence of insolvency proceedings, specified judgments, specified cross-defaults or specified revocations. Upon the
occurrence of an event of default and in the event of a change of control, BNP Paribas and IMBC Group may accelerate
payments due under the Loan Agreements or terminate the Loan Agreements, which would have an adverse impact on
our business.
9
Future issuances of equity or convertible debt securities may cause dilution to our shareholders and ADS holders,
restrict our operations or require us to relinquish rights to our product candidates.
We expect to finance our cash needs through a combination of revenues from product sales, equity offerings, debt
financings, collaborations, strategic alliances and/or licensing arrangements. To the extent that we raise additional
capital through the sale of equity or convertible debt securities, the ownership interests of our shareholders and ADS
holders will be diluted, and the terms of these securities may include liquidation or other preferences that adversely
affect the rights of ordinary shareholders and ADS holders. Debt financing, if available, may involve agreements that
include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making
capital expenditures or declaring dividends.
If we raise funds through further collaborations, strategic alliances or licensing arrangements with third parties, we may
have to relinquish valuable rights to our future revenue streams, research programs or product candidates or to grant
licenses on terms that may not be favorable to us. If we are unable to raise funds through equity or debt financings when
needed, we may be required to delay, limit, reduce or terminate our research and product development or current or
future commercialization efforts or grant rights to develop and market product candidates that we would otherwise
prefer to develop and market ourselves.
Our indebtedness could limit cash flow available for our operations, expose us to risks that could adversely affect our
business, financial condition and results of operations and impair our ability to satisfy our obligations under our
indebtedness.
On July 30, 2024, we issued $426.1 million (A$650.0 million) principal amount of 2.375% unsecured convertible bonds
due 2029 ("the Convertible Bonds") on the SGX. Additionally, as of December 31, 2025, we had $10.3 million of other
indebtedness. We may also incur additional indebtedness to meet future financing needs. Our indebtedness could have
significant negative consequences for our security holders and our business, results of operations and financial condition
by, among other things:
•increasing our vulnerability to adverse economic and industry conditions;
•limiting our ability to obtain additional financing;
•requiring the dedication of a portion of our cash flow from operations to service our indebtedness, which would
reduce the amount of cash available for other purposes;
•limiting our flexibility to plan for, or react to, changes in our business;
•diluting the interests of our existing shareholders as a result of issuing ordinary shares upon conversion of the
Convertible Bonds; and
•placing us at a possible competitive disadvantage with competitors that are less leveraged than us or have better
access to capital.
Our ability to pay the principal of or interest on the Convertible Bonds or to make cash payments in connection with any
conversion of the Convertible Bonds depends on our future performance, which is subject, in part, to economic, financial,
competitive and other factors beyond our control. Our business may not generate cash flow from operations in the future
sufficient to service the Convertible Bonds or other future indebtedness and make necessary capital expenditures.
If we are unable to redeem the Convertible Bonds for cash when required, or repay the Convertible Bonds when due
at maturity, we may need to seek alternative financing arrangements, which could impose restrictions on our
operations and business.
On July 30, 2024, we completed our issuance of the Convertible Bonds to institutional and professional investors on the
SGX outside of the U.S. The Convertible Bonds mature on July 30, 2029, unless redeemed, repurchased, or converted in
accordance with their terms.
Subject to the satisfaction of conditions in the trust deed, we have the right at our option to redeem all of the bonds on
or after August 13, 2027 if (i) the closing price of our ordinary shares on the ASX exceeds 130% of the then-applicable
conversion price for at least 20 trading days, whether consecutive or not, during any consecutive 30 trading day period
or (ii) conversion rights have been exercised in respect of 85% or more in principal amount of the Convertible Bonds.
We may be required to redeem the Convertible Bonds prior to the maturity date in certain circumstances. Upon the
occurrence of an event constituting a change of control or the delisting of our ordinary shares on the ASX, each
bondholder will have the right under the trust deed governing the Convertible Bonds to require us to redeem all or some
of such bondholder’s Convertible Bonds at their principal amount, together with accrued but unpaid interest. We are also
required under the trust deed to redeem the Convertible Bonds on July 30, 2027 at the option of each holder at their
principal amount, together with accrued but unpaid interest.
We may not be able to redeem all or any of such Convertible Bonds or pay all or any amounts due upon conversions
thereof if we do not have sufficient funds to do so. Non-payment of any principal or interest payable with respect to the
10
Convertible Bonds would constitute an event of default under the trust deed governing the Convertible Bonds. Upon the
occurrence of an event of default, the full principal amount, together with accrued but unpaid interest, of the Convertible
Bonds then outstanding will become due and payable. A default under the trust deed could also lead to a default under
agreements governing any of our indebtedness outstanding at the time. If we are unable to redeem the Convertible
Bonds at maturity or upon the occurrence of certain events specified by the trust deed governing the Convertible Bonds,
we may need to seek alternative financing arrangements, which could impose restrictions on our operations and
business. We cannot assure you that such alternative financing will be available to us on acceptable terms, if at all.
Servicing the Convertible Bonds will require a significant amount of cash, and we may not have sufficient cash flow
from our business to make payments on the Convertible Bonds.
Our ability to make scheduled payments of the principal of, to pay interest on or to refinance the Convertible Bonds
depends on our future performance, which is subject to economic, financial, competitive and other factors beyond our
control. Our business may not generate sufficient cash flow from operations in the future to service the Convertible
Bonds. If we are unable to generate sufficient cash flow, we may be required to adopt one or more alternatives, such as
selling assets, restructuring debt or obtaining additional share capital on terms that may be unfavorable to us or highly
dilutive. Our ability to refinance our indebtedness will depend on the capital markets and our financial condition at the
time we seek to refinance such indebtedness. We may not be able to engage in any of these activities or engage in these
activities on desirable terms, which could result in a default on our debt obligations.
We have engaged and plan to engage in various acquisitions and strategic partnerships in the future. If we engage in
acquisitions or strategic partnerships, this may increase our capital requirements, dilute our shareholders and ADS
holders, cause us to incur debt or assume contingent liabilities and subject us to other risks.
We have engaged and plan to continue to engage in various acquisitions and strategic partnerships in the future,
including licensing or acquiring complementary products, intellectual property rights, technologies or businesses. Any
acquisition or strategic partnership may entail numerous risks, including:
•increased operating expenses and cash requirements;
•the assumption of indebtedness or contingent liabilities;
•the issuance of our equity securities which would result in dilution to our shareholders and ADS holders;
•assimilation of operations, intellectual property, products and product candidates of an acquired company,
including difficulties associated with integrating new personnel;
•the diversion of our management’s attention from our existing product programs and initiatives in pursuing such an
acquisition or strategic partnership;
•retention of key employees, the loss of key personnel and uncertainties in our ability to maintain key business
relationships;
•risks and uncertainties associated with the other party to such a transaction, including the prospects of that party
and their existing products or product candidates and regulatory approvals; and
•our inability to generate revenue from acquired intellectual property, technology and/or products sufficient to meet
our objectives or even to offset the associated transaction and maintenance costs.
RLS may have liabilities that are not known to us and the indemnities in the purchase agreement may not offer
adequate protection.
As part of the acquisition, we have agreed to assume certain liabilities of RLS. In addition, there may be liabilities that we
failed or were unable to discover in the course of performing due diligence investigations, including with respect to
decommissioning and decontamination ("D&D"), requirements with respect to the closing of facilities that use and
produce radioactive materials. Also, we may not have correctly assessed the significance of certain liabilities and assets
identified in the course of our due diligence. Any such liabilities, individually or in the aggregate, could have a material
adverse effect on our business, financial condition and results of operations. As we integrate RLS into our operations, we
may learn additional information about the entity, such as unknown or contingent liabilities and issues relating to
compliance with applicable laws, that could potentially have an adverse effect on our business, financial condition and
results of operations.
We may not be able to effectively integrate the businesses that we have acquired and/or may acquire in the future.
Our ability to realize the anticipated benefits of acquisitions we have completed and/or may complete in the future,
including the completed acquisitions of RLS (USA) Inc. ("RLS"), QSAM Biosciences, Inc. ("QSAM"), ARTMS Inc. ("ARTMS"),
IsoTherapeutics Group, LLC, ("IsoTherapeutics"), and certain assets of ImaginAb, Inc., ("ImaginAb") will depend on our
ability to integrate those businesses with our own. The combination of multiple independent businesses is a complex,
costly and time-consuming process and there can be no assurance that we will be able to successfully integrate
businesses into our business, or if such integration is successfully accomplished, that such integration will not be costlier
11
or take longer than presently contemplated. If we cannot successfully integrate and manage the businesses within a
reasonable time, such difficulties or delays could result in the loss of key employees from the acquired businesses, the
disruption of the acquired businesses, inefficiencies, or inconsistencies in standards, controls, information technology
systems, procedures and policies, and we may not be able to realize the potential and anticipated benefits of such
acquisitions, which could have a material adverse effect on our business, financial position, and results of operations. We
face numerous risks relating to the integrated of acquired businesses, including:
•the inability to integrate effectively the operations, products, technologies and personnel of the acquired companies
(some of which are in diverse geographic regions) and achieve expected synergies;
•the potential disruption of existing business and diversion of management’s attention from day-to-day operations;
•the inability to maintain uniform standards, controls, procedures and policies;
•the need or obligation to divest portions of the acquired companies to satisfy regulatory requirements;
•the potential failure to identify material problems and liabilities during due diligence review of acquisition targets;
•the potential failure to obtain sufficient indemnification rights to fully offset possible liabilities associated with
acquired businesses; and
•the challenges associated with operating in new product segments and/or geographic regions.
The failure to maintain our licenses and realize their benefits may harm our business.
We have acquired and in-licensed certain of our technologies from third parties. We may in the future acquire, in-license
or invest in additional technology that we believe would be beneficial to our business. We are subject to a number of
risks associated with our acquisition, in-license or investment in technology, including the following:
•diversion of financial and managerial resources from existing operations;
•successfully negotiating a proposed acquisition, in-license or investment in a timely manner and at a price or on
terms and conditions favorable to us;
•successfully combining and integrating a potential acquisition into our existing business to fully realize the benefits
of such acquisition;
•the impact of regulatory reviews on a proposed acquisition, in-license or investment; and
•the outcome of any legal proceedings that may be instituted with respect to the proposed acquisition, in-license or
investment.
If we fail to properly evaluate potential acquisitions, in-licenses, investments or other transactions associated with the
creation of new R&D programs or the maintenance of existing ones, we might not achieve the anticipated benefits of any
such transaction, we might incur costs in excess of what we anticipate, and management resources and attention might
be diverted from other necessary or valuable activities.
Risks Related to Commercialization and Product Development
Our business is substantially dependent on the commercial success of our Commercial Products and our product
candidates, if approved. If we are unable to successfully commercialize our Commercial Products as currently
approved or to successfully obtain regulatory approvals to commercialize our other product candidates, our
business, financial condition and results of operations will be materially harmed.
Our business and our ability to generate product revenue from the sales of diagnostic imaging agents and therapies that
treat cancer and other diseases depend on continued commercialization of our Commercial Products in the jurisdictions
in which they have received marketing authorization. We are also developing our Commercial Products for additional
indications. We are currently pursuing marketing authorizations for our Commercial Products, either directly or in
collaboration with regional commercial partners, in a number of additional jurisdictions. We believe that obtaining these
additional regulatory approvals and successfully developing our Commercial Products for additional potential indication
will be important to reach the full potential utilization of our Commercial Products and failure to do so could have a
material adverse effect on our business.
Our long-term prospects also depend on our ability to obtain regulatory approval for additional imaging and therapeutic
product candidates. Regulatory approvals are subject to changing standards from time to time and the timing to obtain
the required regulatory approvals is subject to many factors, some of which may be outside our control. For example,
regulatory agencies may face resource constraints, causing delays in the review process, and there is no guarantee that
the regulators are bound by any product development or regulatory advice offered earlier in the review process. In May
2024, we completed our submission of a biologics license application ("BLA"), to the FDA for TLX250-Px for the
12
characterization of renal masses as clear cell renal cell carcinoma ("ccRCC"). In July 2024, the FDA declined to review
the BLA and issued a Refuse to File ("RTF"), determination. A RTF determination is a response from the FDA following its
preliminary review, communicating the FDA’s determination that the application does not include all pertinent information
and data. The denial of acceptance for filing was based on a filing concern related to demonstrating adequate sterility
assurance during dispensing of TLX250-Px in the radiopharmacy production environment. In December 2024 we
resubmitted our BLA to the FDA for TLX250-Px, and the FDA accepted our application in February 2025. In August 2025,
the FDA issued a Complete Response Letter ("CRL"), citing deficiencies relating to the Chemistry, Manufacturing, and
Controls ("CMC") package. The FDA requested additional data to establish comparability between the drug product used
in the ZIRCON Phase 3 clinical trial and the scaled-up manufacturing process intended for commercial use. Additionally,
the FDA documented notices of deficiency (Form 483) issued to two third-party manufacturing and supply chain
partners that required remediation prior to resubmission. In December 2025 we participated in a Type A meeting with the
FDA to align on plans to address CMC deficiencies cited, and in January 2026 we participated in an additional Type A
meeting to align on plans to address comparability between the clinical drug product and that intended for commercial
use. Following these meetings, Telix believes it has alignment with the Agency on key resubmission aspects. While we
believe that the planned remediation of the BLA for TLX250-Px will meet FDA requirements, there can be no assurance
that, once re-submitted, the FDA will accept our BLA for review, or approve TLX250-Px.
In August 2024, we submitted a NDA for TLX101-Px for the characterization of progressive or recurrent glioma from
treatment related changes in both adult and pediatric patients. In October 2024, the FDA accepted the New Drug
Application ("NDA"), and granted priority review and assigned a Prescription Drug User Fee Act ("PDUFA"), goal date of
April 26, 2025. In April 2025, the FDA issued a CRL, stating that additional confirmatory clinical evidence was required to
progress the application. Following engagement with the FDA, including a successful Type A meeting, in September 2025
we reached agreement with the FDA regarding our NDA and are finalizing our resubmission package. There can be no
assurance that the FDA will accept our resubmitted NDA for review, or approve TLX101-Px.
In February 2026, we submitted a Marketing Authorization Application ("MAA") for TLX101-Px in Europe, with France
acting as the Reference Member State ("RMS"). The French National Agency for Medicines and Health Products Safety
("ANSM") in its capacity as RMS is responsible for coordinating and leading the scientific evaluation of the dossier, in
collaboration with the Concerned Member States, nominated by Telix and representing the major European markets for
Telix’s brain cancer imaging product. There can be no assurance that the MAA will be validated, or that marketing
authorizations for TLX101‑Px will ultimately be granted.
Any adverse action by the FDA with respect to the BLA or NDAs, could delay our planned commercial development
timelines or could prevent us from commercializing these product candidates. If the FDA determines that our
submissions and the data supporting the submissions are not sufficient to support approval in these indications, we may
be required to conduct an additional clinical trial or trials, which would increase our costs and delay the program. Any
such delay or other adverse impact could have a material adverse effect on our business.
We have not submitted any applications for regulatory approval or obtained regulatory approval for any of our
therapeutic product candidates. Our most advanced therapeutic candidate, TLX591-Tx (lutetium (Lu177) rosopatamab
tetraxetan), is a lutetium-labeled radio antibody-drug conjugate ("rADC"), which we are evaluating in a two-part Phase 3
clinical trial in patients with advanced prostate cancer, called ProstACT Global. We dosed the first patient in this clinical
trial in November 2023 in Australia. We received authorization to conduct Part 1 of the trial in the U.S. in April 2024 and
completed target enrollment of 30 patients in August 2025. We dosed the first patient in ProstACT Global Part 2
(randomized treatment expansion) in Australia in December 2025. We cannot be certain that TLX591-Tx, or any of our
clinical trials of our other therapeutic product candidates, will generate safety and efficacy data sufficient for regulatory
approval in any jurisdiction.
The commercial success of our Commercial Products and our product candidates, if approved, is dependent on many
factors, some of which are beyond our control, including clinical development, the regulatory submission and approval
process, market access or reimbursement frameworks, potential threats to our intellectual property rights and the
manufacturing, marketing and sales efforts. If we are unable to continue to commercialize our Commercial Products or to
develop, receive regulatory approval for and successfully commercialize our product candidates, or experience delays as
a result of any of these factors or otherwise, our business and results of operations could be substantially harmed.
Clinical development is a lengthy and expensive process, with uncertain timelines and outcomes. If clinical trials of
our product candidates fail to demonstrate safety and efficacy to the satisfaction of regulatory authorities or do not
otherwise produce positive results, we may incur additional costs or experience delays in completing, or ultimately
be unable to complete, the development and commercialization of such product candidates, if approved.
Our long-term success depends in large part on our ability to continue to successfully develop additional product
candidates in imaging and therapeutic indications. Clinical testing is expensive, time consuming, difficult to design and
implement, and is inherently uncertain as to outcome. Clinical failure can occur at any stage of the clinical development
process and, therefore, the outcome of preclinical studies and early-stage clinical trials may not be predictive of the
success of later stage clinical trials. Furthermore, the failure of any product candidates to demonstrate safety and
efficacy in any clinical trial could negatively impact the perception of our company or our products and/or cause the FDA
or other regulatory authorities to require additional testing before any of our product candidates are approved.
13
We may experience numerous unforeseen events during, or as a result of, clinical trials that could delay or prevent our
ability to receive regulatory approval of our product candidates, including, but not limited to, the following:
•delays or failure to reach agreement with regulatory authorities on a trial design or the receipt of feedback requiring
us to modify the design of our clinical trials, perform additional or unanticipated clinical trials to obtain approval or
alter our regulatory strategy;
•clinical trials of our product candidates may produce negative or inconclusive results or other patient safety
concerns, including undesirable adverse events or other unexpected characteristics, and we may decide, or
regulatory authorities may require us, to conduct additional clinical trials, suspend ongoing clinical trials or abandon
product development programs, including as a result of a finding that the participants are being exposed to
unacceptable health risks;
•enrollment in our clinical trials may be slower than we anticipate or we may not be able to enroll the number of
patients that we expect, including as a result of competition with other ongoing clinical trials for the same
indications as our product candidates or because the patient population may be limited for orphan indications;
•regulators may revise the requirements for approving our product candidates, even after providing a positive
opinion on or otherwise reviewing and providing comments on a clinical trial protocol, or such requirements may not
be as we anticipate;
•delays or failure in obtaining the necessary authorization from regulatory authorities or institutional review boards to
permit us or our investigators to commence a clinical trial, conduct a clinical trial at a prospective trial site, or the
suspension or termination of a clinical trial once commenced;
•delays or failure to reach agreement on acceptable terms with prospective clinical trial sites or contract research
organizations ("CROs");
•delays in manufacturing, testing, releasing, validating or importing/exporting sufficient stable quantities of our
product candidates for use in clinical trials or the inability to do any of the foregoing;
•the number of patients required for clinical trials of our product candidates may be larger than we anticipate or
participants may drop out of these clinical trials at a higher rate than we anticipate;
•our third-party contractors, including manufacturers or CROs, may fail to comply with regulatory requirements,
perform effectively, or meet their contractual obligations to us in a timely manner, or at all;
•we or our investigators might be found to be non-compliant with regulatory requirements;
•the cost of clinical trials of our product candidates may be greater than we anticipate;
•the supply or quality of our product candidates or other materials necessary to conduct clinical trials may be
insufficient or inadequate;
•regulators or institutional review boards/ethics committees may not authorize us or our investigators to commence
a clinical trial or conduct a clinical trial at a prospective trial site;
•imposition of a temporary or permanent clinical hold by regulatory authorities for a number of reasons, including
after review of an IND or amendment or equivalent foreign application or amendment, as a result of a new safety
finding that presents unreasonable risk to clinical trial participants, or a negative finding from an inspection of our
clinical trial operations or study sites;
•developments on trials conducted by competitors for related technology that raises FDA or foreign regulatory
authority concerns about risk to patients of the technology broadly, or if the FDA or a foreign regulatory authority
finds that the investigational protocol or plan is clearly deficient to meet its stated objectives;
•occurrence of adverse events associated with the product candidate that are viewed to outweigh its potential
benefits, or occurrence of adverse events in trial of the same class of agents conducted by other companies;
•any partners or collaborators that help us conduct clinical trials may face any of the above issues, and may conduct
clinical trials in ways they view as advantageous to them but that are suboptimal for us; and
•negative impacts resulting from infectious disease epidemics or pandemics, including impacts to healthcare
systems and our trial sites’ ability to conduct trials.
If we are required to conduct additional clinical trials or other testing of our product candidates beyond those that we
currently contemplate or are unable to successfully complete clinical trials of our product candidates or other testing, on
14
a timely basis or at all, and/or if the results of these trials or tests are not positive or are only modestly positive or if there
are safety concerns, we may:
•be delayed in obtaining, or not obtain at all, regulatory approval for the indication or product candidate;
•obtain regulatory approval in some countries and not in others;
•obtain approval for indications or patient populations that are not as broad as intended or desired;
•obtain approval with labeling that includes significant use or distribution restrictions or safety warnings, including
boxed warnings;
•be subject to additional post-marketing testing requirements; or
•have the product removed from the market after obtaining regulatory approval.
Further, we do not know whether clinical trials will begin as planned, will need to be restructured or will be completed on
schedule, or at all. Significant clinical trial delays also could shorten any periods during which we may have the exclusive
right to commercialize our products, allow our competitors to bring products to market before we do or impair our ability
to successfully commercialize our products, which would harm our business and results of operations. In addition, many
of the factors that cause, or lead to, clinical trial delays may ultimately lead to the denial of regulatory approval of our
product candidates.
If we experience delays or difficulties in enrolling patients in our ongoing or planned clinical trials, our receipt of
necessary regulatory approval could be delayed or prevented.
We may not be able to initiate or continue our ongoing or planned clinical trials for our product candidates if we are
unable to identify and enroll a sufficient number of eligible patients to participate in these trials as required by the FDA or
other applicable foreign regulator. In addition, some of our competitors may have planned or ongoing clinical trials or
expanded access programs for approved and/or investigational products that would treat the same patients as our
therapeutic product candidates, and patients who would otherwise be eligible for our clinical trials may instead enroll in
our competitors’ clinical trials or expanded access programs. Patient enrollment is also affected by other factors,
including:
•severity of the disease under investigation;
•our ability to recruit clinical trial investigators of appropriate competencies and experience;
•the incidence and prevalence of our target indications;
•clinicians’ and patients’ awareness of, and perceptions as to the potential advantages and risks of our product
candidates in relation to other available therapies, including any new products that may be approved for the
indications we are investigating;
•invasive procedures required to enroll patients and to obtain evidence of the product candidate’s performance
during the clinical trial;
•availability and efficacy of approved medications for the disease under investigation;
•eligibility criteria defined in the protocol for the trial in question;
•the ability of our companion diagnostics to identify patients;
•the size of the patient population required for analysis of the trial’s primary endpoints;
•efforts to facilitate timely enrollment in clinical trials;
•whether we are subject to a partial or full clinical hold on any of our clinical trials;
•reluctance of physicians to encourage patient participation in clinical trials;
•the ability to monitor patients adequately during and after treatment;
•our ability to obtain and maintain patient consents; and
•proximity and availability of clinical trial sites for prospective patients.
Our inability to enroll and retain a sufficient number of patients for our clinical trials would result in significant delays or
may require us to abandon one or more clinical trials altogether. Enrollment delays in our clinical trials may result in
15
increased development costs, which would cause the value of our company to decline and limit our ability to obtain
additional financing.
Serious adverse events related to our Commercial Products or our product candidates may delay or prevent their
regulatory approval, cause us to suspend or discontinue clinical trials or abandon further development, limit the
commercial value of approved indications or result in significant negative financial consequences following any
regulatory approval.
If our Commercial Products or any of our product candidates are associated with undesirable adverse events or have
characteristics that are unexpected in clinical trials or following approval and/or commercialization, we may need to
abandon or limit their development or limit marketing to certain uses or subpopulations in which the undesirable adverse
events or other characteristics are less prevalent, less severe or more acceptable from a risk-benefit perspective.
Adverse events in our clinical trials to date have been generally predictable and typically manageable, with frequency
and severity for adverse events applicable to imaging less than for therapy product candidates. The most common
adverse events for Illuccix in clinical trials were nausea, diarrhea, and dizziness. The most common adverse events
arising in the Phase 3 ZIRCON clinical trial of 300 patients dosed with TLX250-Px were mild and non-serious, including
nausea, procedural pain and headache. The most common severe adverse events were post-procedural hemorrhage (six
events), urinary retention (three events), hypertension (three events), pyelonephritis (two events), anemia (two events),
and syncope (two events). For TLX101-Px there have been two events reported to date in an ongoing clinical trial, which
are injection site reaction and nausea, both mild and non-serious.
With respect to our therapeutic product candidates, our most clinically advanced therapeutic, TLX591-Tx, has been
evaluated in 242 patients across eight Phase 1 and 2 trials, including the Phase 1 ProstACT SELECT trial. In Cohort 2 of
ProstACT SELECT (25 participants), treatment emergent adverse events ("TEAE") rates were: Grade 3 thrombocytopenia
(16%, 4/25), grade 3 neutropenia (28%, 7/25), grade 4 thrombocytopenia (20%, 5/25) and grade 4 neutropenia (4%,
1/25). Six patients received intervention in the form of platelet or red blood cell transfusion(s), or both. Early-stage trial
results should be interpreted with caution and efficacy outcomes should be evaluated for statistical and clinical
significance in a larger Phase 3 randomized controlled trial. TLX591-Tx is presently being investigated in Telix's Phase 3
ProstACT Global trial.
The occurrence of adverse events in either our clinical trials or following regulatory approval could result in a more
restrictive label for any product candidates approved for marketing or could result in the delay or denial of approval to
market any product candidates by the FDA or comparable foreign regulatory authorities, which could prevent us from
generating sufficient revenue from product sales or maintaining profitability. Treatment-related adverse effects could
also affect patient recruitment or the ability of enrolled patients to complete the trial, result in potential product liability
claims or cause patients and/or healthcare providers to elect alternative courses of treatment. In addition, these adverse
events may not be appropriately recognized or managed by the treating medical staff. Inadequate training or education
of healthcare professionals to recognize or manage the potential adverse events following treatment with our
Commercial Products or our product candidates, if approved, could result in increased treatment-emergent adverse
events and cause patients to discontinue treatment. Any of these occurrences may harm our business, financial
condition and prospects significantly.
Results of our trials could reveal an unacceptably high severity and prevalence of adverse events. In such an event, our
trials could be suspended or terminated by us or the FDA or comparable foreign regulatory authorities could order us to
cease further development of or deny approval of our product candidates for any or all targeted indications.
Adverse events in the results of trials conducted by our competitors could also cause the FDA or comparable foreign
regulatory authorities to raise concerns regarding our trials and product candidates, and/or impose additional safety and
tolerance procedures on us, which may be costly. Many compounds that initially showed promise in early-stage trials for
treating cancer or other diseases have later been found to cause adverse events that prevented further development of
the compound. If such an event occurs after any of our product candidates are approved and/or commercialized, a
number of potentially significant negative consequences may result, including:
•regulatory authorities may withdraw the approval of such product;
•regulatory authorities may require additional warnings on the label, such as a “black box” warning, precaution or a
contraindication, or issue safety alerts, Dear Healthcare Provider letters, press releases or other communications
containing warnings or other safety information about the product, or impose distribution or use restrictions;
•patients and/or healthcare providers may elect to utilize other treatment options that have or are perceived to have
more tolerable adverse events;
•regulatory authorities may require one or more post-marketing studies;
•we may be required to implement a Risk Evaluation and Mitigation Strategy ("REMS"), or create a medication guide
outlining the risks of such adverse events for distribution to patients;
•additional restrictions may be imposed on the marketing or promotion of the particular product or the manufacturing
processes for the product or any component thereof;
16
•we could be sued and held liable for harm caused to patients;
•the product could become less competitive; and
•our reputation may suffer.
Further, we and our clinical trial investigators currently determine if serious adverse events are product-related in
accordance with scientific practice and current knowledge. The FDA or foreign regulatory authorities may disagree with
our or our clinical trial investigators’ interpretation of data from clinical trials and the conclusion by us or our clinical trial
investigators that a serious adverse event was not product-related. The FDA or foreign regulatory authorities may
require more information related to the safety profile of our Commercial Products or our product candidates, including
additional preclinical or clinical data to support approval, which may cause us to incur additional expenses, delay or
prevent the approval of one of our product candidates, and/or delay or cause us to change our commercialization plans,
or we may decide to abandon the development of the product candidate altogether.
Any of these events could prevent the affected product candidate, if approved, from achieving or maintaining market
acceptance, or could substantially increase costs and expenses of development or commercialization, which could delay
or prevent us from generating sufficient revenue from the sale of our Commercial Products or any other approved
product and harm our business and results of operations.
The results of previous clinical trials may not be predictive of future trial results, and preliminary, interim or top-line
data may be subject to change or qualification based on the complete analyses of data and, therefore, may not be
predictive of the final results of a trial.
Clinical failure can occur at any stage of the clinical development process and, therefore, the outcome of preclinical
studies and early-stage clinical trials may not be predictive of the success of later stage clinical trials. For example,
preliminary, interim or top-line data may be based on unaudited data provided by our clinical trial investigators.
Finalization and cleaning of this data may change the conclusions drawn from this unaudited data provided by our clinical
trial investigators indicating less promising results than we currently anticipate. Further, there can be significant
variability in safety and/or efficacy results between different trials of the same product candidate due to numerous
factors, including changes in trial protocols, differences in size and type of the patient populations, adherence to the
dosing regimen and other trial protocols and the dropout rate among clinical trial participants. We do not know whether
any clinical trials we may conduct will demonstrate consistent or adequate efficacy and safety data sufficient to obtain
regulatory approval to market our product candidates, if approved. Moreover, preclinical and clinical data are often
susceptible to varying interpretations and analyses, and many companies have suffered significant setbacks in late-
stage clinical trials after achieving positive results in earlier development, and we could face similar setbacks.
We may publicly disclose preliminary, interim or top-line data from our clinical trials. Disclosures are based on a
preliminary analysis of then-available data, and the results and related findings and conclusions are subject to change as
further patient data become available and following a more comprehensive review of the data related to the particular
study or trial. For any study that we report preliminary, interim or top-line data, we make assumptions, estimations,
calculations and conclusions as part of our analyses of data. We may not have received or had the opportunity to fully
and carefully evaluate all data, or our conclusions may differ from those of the FDA or other regulatory authorities.
Consequently, the preliminary, interim or top-line data results that we report may differ from future results of the same
studies, or different conclusions or considerations may qualify such results, once additional data have been received and
fully evaluated or based on differing views from regulatory agencies. Preliminary, interim or top-line data also remain
subject to audit and verification procedures that may result in the final data being materially different from the
preliminary data we previously published. As a result, these early data points should be viewed with caution until the final
data are available. Adverse differences between previous preliminary or interim data and future interim or final data could
significantly harm our business.
In addition, the information we choose to publicly disclose regarding a particular study or clinical trial is typically selected
from a more extensive amount of available information. Furthermore, we may report interim analyses of only certain
endpoints rather than all endpoints. Investors may not agree with what we determine is the material or otherwise
appropriate information to include in our disclosure, and any information we determine not to disclose may ultimately be
deemed significant with respect to future decisions, conclusions, views, activities or otherwise regarding a particular
product, product candidate or our business.
If the preliminary, interim or top-line data that we report differ from final results, or if others, including regulatory
authorities, disagree with the conclusions reached, our ability to obtain approval for and commercialize our product
candidates, if approved, may be harmed, which could harm our business, operating results, prospects, or financial
condition.
Our approach to the discovery and development of therapeutic product candidates represents a novel approach to
radiation therapy, which creates significant and potentially unpredictable challenges for us.
Our success depends on the successful development of our therapeutic product candidates, which are designed to treat
solid tumors using a novel approach to radiation therapy. There are currently few approved radiopharmaceutical
17
therapeutic products. In addition, there has been limited historical clinical trial experience, generally, for the development
of radiopharmaceutical therapeutics. As a result, the design and conduct of clinical trials for these drugs is uncertain and
subject to increased risk.
While the use of external beam radiation as a therapy for cancers has existed for decades, the use of systemic delivery
of targeted radiopharmaceuticals in general is relatively new, including for both beta- and alpha-emitting therapies. It is
difficult to accurately predict the challenges we may incur for our therapeutic product candidates as they proceed
through clinical trials. In addition, assessments of the long-term safety of targeted beta- and alpha-emitting isotope
therapies have been limited, and there may be long-term effects from treatment with our therapeutic product candidates
that we cannot predict at this time.
Any difficulties or delays in the commencement or completion, or termination or suspension, of our ongoing or
planned clinical trials could result in increased costs to us, delay or limit our ability to generate revenue and adversely
affect our commercial prospects.
Before obtaining marketing approval from regulatory authorities for our product candidates, we must conduct extensive
clinical trials to demonstrate the safety and efficacy of the product candidates in humans. Before we can initiate clinical
trials for any future product candidates, we must submit the results of preclinical studies to the FDA or comparable
foreign regulatory authorities along with other information, including information about product candidate chemistry,
manufacturing and controls and our proposed clinical trial protocol, as part of an IND or similar regulatory filing required
for authorization to proceed with clinical development. The FDA or comparable foreign regulatory authorities may require
us to conduct additional preclinical studies for any product candidate before it allows us to initiate clinical trials under any
IND or similar regulatory filing, which may lead to delays and increase the costs of our preclinical development programs.
Moreover, even if we commence clinical trials, issues may arise that could cause regulatory authorities to suspend or
terminate such clinical trials. Any such delays in the commencement or completion of our ongoing or planned clinical
trials for our product candidates could significantly affect our product development timelines and product development
costs.
We do not know whether our planned and ongoing trials will begin on time or be completed on schedule, if at all. The
commencement, data readouts and completion of clinical trials can be delayed for a number of reasons, including delays
related to:
•obtaining regulatory authorizations to commence a trial or reaching a consensus with regulatory authorities on trial
design;
•the FDA or comparable foreign regulatory authorities disagreeing as to the design or implementation of our clinical
studies;
•any failure or delay in reaching an agreement with CROs and clinical trial sites, the terms of which can be subject to
extensive negotiation and may vary significantly among different CROs and trial sites;
•obtaining approval from one or more institutional review boards ("IRBs");
•IRBs or ethics committees refusing to approve, suspending or terminating the trial at an investigational site,
precluding enrollment of additional subjects, or withdrawing their approval of the trial;
•changes to the clinical trial protocol;
•delays in identifying, recruiting and training suitable clinical investigators;
•clinical sites deviating from the trial protocol or dropping out of a trial;
•manufacturing sufficient quantities of our product candidates for use in clinical trials;
•subjects failing to enroll or remain in our trials at the rate we expect, or failing to return for post-treatment follow-
up, including subjects failing to remain in our trials due to movement restrictions, health reasons or otherwise
resulting from ongoing or future public health or geopolitical concerns;
•subjects choosing alternative treatments for the indications for which we are developing our therapeutic product
candidates, or participating in competing clinical trials;
•lack of adequate funding to continue the clinical trial or incurring greater costs than we anticipate;
•subjects experiencing severe or serious unexpected treatment-emergent adverse events;
•occurrence of serious adverse events in trials of the same class of agents conducted by other companies;
•selection of clinical endpoints that require prolonged periods of clinical observation or extended analysis of the
resulting data;
18
•failure of a facility manufacturing our product candidates or any of their components to produce clinical trial
materials in accordance with current good manufacturing practice requirements("cGMP"), regulations (and similar
foreign requirements) or other applicable requirements;
•a facility manufacturing our product candidates or any of their components being ordered by the FDA or
comparable foreign regulatory authorities to temporarily or permanently shut down due to violations of cGMP
regulations (and similar foreign requirements) or other applicable requirements, or infections or cross-
contaminations of product candidates in the manufacturing process;
•any transfer of manufacturing processes to alternate facilities or any other changes to our manufacturing process
that may be necessary or desired;
•third-party clinical investigators losing the licenses or permits necessary to perform our clinical trials, not
performing our clinical trials on our anticipated schedule or consistent with the clinical trial protocol, good clinical
practice ("GCP"), requirements or other regulatory requirements;
•third-party contractors not performing data collection or analysis in a timely or accurate manner; or
•third-party clinical investigators or becoming debarred or suspended or otherwise penalized by the FDA or other
government or regulatory authorities for violations of regulatory requirements, in which case we may need to find a
substitute contractor, and we may not be able to use some or all of the data produced by such clinical investigators
or contractors in support of our marketing applications.
We could encounter delays if a clinical trial is suspended or terminated by us, by the IRBs of the institutions in which such
trials are being conducted, by a Data Safety Monitoring Board for such trial or by the FDA or comparable foreign
regulatory authorities. Such authorities may impose such a suspension or termination due to a number of factors,
including failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols,
inspection of the clinical trial operations or trial site by the FDA or comparable foreign regulatory authorities resulting in
the imposition of a clinical hold, unforeseen safety issues or adverse events, failure to demonstrate a benefit from using a
drug or diagnostic, changes in governmental regulations or administrative actions or lack of adequate funding to continue
the clinical trial. In addition, changes in regulatory requirements and policies may occur, and we may need to amend
clinical trial protocols to comply with these changes. Amendments may require us to resubmit our clinical trial protocols
to IRBs or ethics committees for reexamination, which may impact the costs, timing or successful completion of a clinical
trial.
If we experience delays in the completion of, or termination of, any clinical trial of our product candidates, the
commercial prospects of our product candidates will be harmed, and our ability to generate product revenues from any
of these product candidates, if approved, will be delayed. Any delays in completing our clinical trials will increase our
costs, slow down our product candidate development and approval process and jeopardize our ability to commence
product sales and generate revenues if the product candidate is approved. Such delays could also shorten any period
during which we may have the exclusive right to commercialize our product candidates, if approved, and our competitors
may be able to bring products to market before we do, and the commercial viability of our product candidates could be
significantly reduced. In addition, many of the factors that cause, or lead to, the termination or suspension of, or a delay
in the commencement or completion of, clinical trials may also ultimately lead to the denial of regulatory approval of a
product candidate. Any of these occurrences may harm our business, financial condition and prospects significantly.
We may find it difficult to enroll patients in our clinical trials. If we encounter difficulties enrolling subjects in our
clinical trials, our clinical development activities could be delayed or otherwise adversely affected.
Patient enrollment is a significant factor in the timing of clinical trials, and the timing of our clinical trials depends, in part,
on the speed at which we can recruit patients to participate in our trials, as well as completion of required follow-up
periods. We may not be able to initiate or continue clinical trials for our product candidates if we are unable to identify
and enroll a sufficient number of eligible patients to participate in these trials as required by the FDA or similar regulatory
authorities outside the U.S. Subject enrollment is affected by many factors including the size and nature of the patient
population, the severity of the disease under investigation, the availability and efficacy of approved drugs and
diagnostics for the disease under investigation, the proximity of patients to clinical sites, the eligibility and exclusion
criteria for the trial, the design of the clinical trial, the risk that enrolled patients will not complete a clinical trial, our ability
to recruit clinical trial investigators with the appropriate competencies and experience, patient referral practices of
physicians, the ability to monitor patients adequately during and after treatment, competing clinical trials and clinicians’
and patients’ perceptions as to the potential advantages and risks of the product candidate being studied in relation to
other available therapies, including any new products that may be approved for the indications we are investigating as
well as any product candidates under development.
We will be required to identify and enroll a sufficient number of subjects for each of our clinical trials. The potential
patient populations for our clinical trials may be narrow, and we may experience difficulties in identifying and enrolling a
sufficient number of patients in our clinical trials. We may not be able to initiate or continue clinical trials if we are unable
to locate a sufficient number of eligible subjects to participate in the clinical trials required by the FDA or comparable
foreign regulatory authorities.
19
Other pharmaceutical or biotechnology companies targeting the same diseases and intended uses as our product
candidates are recruiting for their clinical trials from these patient populations, which may make it more difficult to fully
enroll our clinical trials. Our inability to enroll a sufficient number of subjects for any of our future clinical trials would
result in significant delays or may require us to abandon one or more clinical trials altogether. In addition, the process of
finding eligible subjects may prove costly.
Moreover, we rely on CROs and clinical trial sites to ensure proper and timely conduct of our clinical trials and, while we
intend to enter into agreements governing their services, we will have limited influence over their actual performance. We
cannot assure you that our assumptions used in determining expected clinical trial timelines are correct or that we will
not experience delays in enrollment, which would result in the delay of completion of such trials beyond our expected
timelines.
Due to their radioactive nature, our Commercial Products and our product candidates have time-limited stability, and
as a result, we may encounter difficulties with fulfillment and logistics.
The radioactive components of our Commercial Products and our product candidates have short shelf lives due to their
half lives, which refers to the time it takes for the radioactivity to decrease by 50%. Post addition of the radioactive
isotope, radioactivity decay reduces the shelf life of our Commercial Products and our product candidates, which
requires us to manufacture and deliver our Commercial Products and our product candidates for use in clinical trials to
patients in a timely manner.
Our products and product candidates are commonly manufactured as a cold-kit, namely our Commercial Products,
enabling longer shelf storage of between 12-24 months prior to radiolabeling for specific patient administration on an as-
needed basis. As such, our Commercial Products and our product candidates must be radiolabeled on an as-needed
basis, and shipped almost immediately thereafter. Because of this, specific radiolabeled patient doses of our Commercial
Products or our product candidates cannot be “stock-piled” and stored for even a small number of days ahead of
shipment, we or any third-party pharmacy network or hospital must be able to radiolabel them on an as-needed rolling
basis. Any delay, even if seemingly insignificant, could result in an immediate and substantial impact on our ability to
deliver our Commercial Products or product candidates to patients. Any significant delays in delivering our Commercial
Products or our product candidates to patients could damage our reputation and result in deviations from our clinical trial
protocols, which in turn could affect our ability to advance the clinical development of our current and future product
candidates on a timely basis, or at all. In addition, we currently substantially rely on our third-party radiopharmacy
partners for the production of our Commercial Products for commercial supply in the U.S. We cannot be sure that these
manufacturers will be able to meet our demand for our Commercial Products on a timely basis.
With respect to our product candidates, as we continue to scale our operations and enroll larger clinical trials, and
prepare for potential commercialization, if marketing authorization is obtained, we will need to scale our shipping abilities.
Labor disputes, government restrictions, work stoppages, pandemics, derailments, damage or loss events, adverse
weather conditions, other events beyond our control could interrupt or delay transportation, which could result in the loss
or damage of our Commercial Products or any product candidates with similar stabilization restrictions. We have
insurance which covers material loss or damage to Illuccix and/or Gozellix while in partner control or during transit,
subject to customary insurance limitations and restrictions. Our insurance may not cover all instances worldwide.
If we or our manufacturers are unable to meet the challenges posed by the time-limitations inherent in the composition of
our Commercial Products or any of our product candidates, it would adversely affect our business, financial condition,
results of operations and prospects.
We may not be successful in our efforts to identify or discover additional product candidates or our decisions to
prioritize the development of certain product candidates over others may later prove wrong.
Part of our strategy involves identifying and developing product candidates to build a pipeline of product candidates. Our
diagnostic and therapeutic discovery or development efforts may not be successful in identifying compounds that are
useful in diagnosing or treating cancer or other diseases. Our research programs may initially show promise in identifying
potential product candidates, yet fail to yield product candidates for clinical development for a number of reasons,
including:
•the research methodology used may not be successful in identifying potential product candidates;
•potential product candidates may, on further study, be shown to have harmful adverse events or other
characteristics that indicate that they are unlikely to be products that will receive regulatory approval and/or
achieve market acceptance; or
•potential product candidates may not be effective in treating their targeted diseases or yield clinically significant
outcomes.
We are currently advancing multiple imaging and therapeutic product candidates in clinical development, which may
create a strain on our limited human and financial resources. As a result, we may not be able to provide sufficient
resources to any single product candidate to permit the successful development and commercialization of such product
candidate, if approved, which could result in material harm to our business. Further, we have limited financial and
20
managerial resources, and we can only focus our research programs on developing product candidates for certain
indications.
As a result, we may forego or delay pursuit of opportunities with other product candidates or the same product
candidate for other indications that later prove to have greater commercial potential. Our resource allocation decisions
may cause us to fail to capitalize on viable commercial products or profitable market opportunities. Our spending on
current and future research and development programs and product candidates for specific indications may not yield any
commercially viable products. If we do not accurately evaluate the commercial potential or target market for a particular
product candidate, we may relinquish valuable rights to that product candidate through collaboration, licensing or other
royalty arrangements in cases in which it would have been more advantageous for us to retain sole development and
commercialization rights to such product candidate.
Our strategy involves pairing our diagnostic imaging product or product candidates with a complementary
therapeutic product candidate, and we may not be successful in developing both the diagnostic and therapeutic
product candidates that are designed to be paired, which could impact the successful development of both.
In connection with certain targets for which we are developing drug or biological candidates for treatment use, we are
developing diagnostic imaging agents to help inform whether a particular patient’s disease condition is appropriate for
treatment with our drug or biological candidate. For example, we are using Illuccix as the paired diagnostic to our
therapeutic product candidate, TLX591-Tx (in addition to Illuccix being previously studied and used in the VISION trial as
a diagnostic for Novartis’ Pluvicto radioligand therapy) and we are developing TLX300-Px as the paired diagnostic to
evaluate the potential utility of TLX300-Tx, and similarly we are developing paired diagnostics for our other therapeutic
product candidate development programs. We may not be successful in developing an appropriate diagnostic imaging
agent or its development may cause a delay or result in expenditure of more funds than we currently anticipate. In
addition, the development of a diagnostic imaging agent will be subject to FDA review and approval, which may be
delayed or not obtained, or require additional development and testing than currently planned. If the FDA considers the
diagnostic imaging agent to be required for the use of the therapeutic product candidate, the FDA may require the
approval of the diagnostic imaging agent before it can approve the therapeutic product candidate. Equivalent foreign
regulatory review and approval would also be required before the product could be supplied for use in patient treatment.
Failure to successfully develop and obtain regulatory approval for a diagnostic imaging agent may delay FDA or foreign
regulatory approval of a drug or biological candidate intended for therapeutic use and delay or adversely affect
commercialization of that drug or biological candidate, if approved, or require us to engineer or identify alternative
solutions to select patients who are most likely to benefit from our drug or biological candidates.
We face substantial competition, which may result in others discovering, developing or commercializing products
before or more successfully than we do.
The discovery, development and commercialization of new diagnostics and therapies is highly competitive, particularly in
the cancer field. We face competition with respect to our Commercial Products and will face competition with respect to
any product candidates that we are developing and may seek to commercialize in the future, from major pharmaceutical
companies, specialty pharmaceutical companies, biotechnology companies, academic institutions and governmental
agencies as well as public and private research institutions worldwide, many of which have significantly greater financial
resources and expertise in research and development, manufacturing, preclinical studies, conducting clinical trials,
obtaining regulatory approvals and marketing approved products than we do.
There are a number of major pharmaceutical, specialty pharmaceutical and biotechnology companies that currently
market and sell diagnostics and therapies and/or are pursuing the development of diagnostics and therapies for the
treatment of cancer and the other disease indications for which we are developing our products and product candidates.
With respect to Illuccix and Gozellix, our main competitors in the U.S. include companies with approved PSMA-PET
diagnostics, including Novartis AG, Lantheus Holdings, Inc. and The Bracco Group (through its Blue Earth Diagnostics
affiliate). Certain academic institutions, like University of California, Los Angeles and University of California, San
Francisco, also hold a license for a commercial PSMA-PET diagnostic. Our main competitors also include companies
developing PSMA imaging agents, including Curium Holding France S.A.S., Clarity Pharmaceuticals Limited, ABX
advanced biochemical compounds GmbH, Isotopia Molecular Imaging Ltd., Itel Group, ITM Isotope Technologies Munich
SE, Five Eleven Pharma Inc., FutureChem Co. Ltd., Radiomedix, Inc., CellBion Co., Ltd., Norroy Biosciences Co. Ltd., HTA,
and Jiangsu Hengrui Pharmaceuticals Company Ltd. Our competitors will also include companies developing other
modalities to localize prostate cancer.
In the kidney and brain cancer imaging fields, there are no approved agents for molecular imaging for ccRCC or glioma.
Our main future competitors in these fields are companies developing agents, including ITM Isotope Technologies
Munich SE, Philogen S.p.A., Precision Molecular, Inc., Norroy Biosciences Co. Ltd., PeptiDream Inc., Five Eleven Pharma
Inc., Novartis AG, The Bracco Group (through its Blue Earth Diagnostics Ltd. Subsidiary), RadioPharm Theranostics
Limited, Curasight A/S, Molecular Targeting Technologies, Inc. ("MTTI"), and BoomRay Pharmaceuticals Co., Ltd.
With respect to our therapeutic product candidates, we consider our most direct competitors to be companies
developing targeted radiopharmaceuticals for the treatment of cancer. There are several companies with approved beta-
based radiopharmaceuticals, including Novartis AG, Sirtex Medical Limited, Boston Scientific Corporation, Acrotech
Biopharma LLC, and Q BioMed Inc. and other companies developing beta-based radiopharmaceuticals, including
Lantheus Holdings, Inc., Eli Lilly and Company Ltd, ITM Isotope Technologies Munich SE, Debiopharm SA, Curium Holding
21
France S.A.S, Clarity Pharmaceuticals Limited, The Bracco Group (through its Blue Earth Therapeutics Ltd. subsidiary),
and Y-mAbs Therapeutics, Inc. The beta emitting isotopes used by these companies include Iodine-131, Lutetium-177,
Strontium-89, Copper-67, and Yttrium-90. A recently approved beta particle-based radiopharmaceutical is Pluvicto,
which was developed by Novartis AG and approved by the FDA in 2022 for the treatment of patients with metastatic
prostate cancer.
There are also several companies developing targeted alpha-based radiopharmaceuticals for the treatment of cancer,
including Bayer AG, Novartis AG, Johnson & Johnson, Abdera Therapeutics Inc., Actinium Pharmaceuticals, Inc., Aktis
Oncology, Inc., Convergent Therapeutics, Inc., AstraZeneca PLC, ITM Isotope Technologies Munich SE, Perspective
Therapeutics, Inc., Eli Lilly & Company Ltd, RadioMedix, Inc., Bristol Myers Squibb Company, and Y-mAbs Therapeutics,
Inc. These companies are targeting a wide range of solid and hematologic malignancies using various alpha-emitting
isotopes, including Radium-223, Lead-212, and Actinium-225. The first and only approved alpha particle-based therapy
is Xofigo (Radium-223), which was developed by Bayer AG and approved in 2013 for the treatment of prostate cancer
with symptomatic bone metastases.
With respect to TLX591-Tx (lutetium (Lu177) rosopatamab tetraxetan), our main competitors include Novartis AG, with
Pluvicto as the only currently approved PSMA-targeted therapy. Our main competitors also include companies
developing PSMA-targeted therapies, including AstraZeneca PLC, Convergent Therapeutics, Inc., Eli Lilly & Company
Ltd., Lantheus Holdings, Inc., Curium Holding SAS, ArtBio, Inc., The Bracco Group (through its Blue Earth Therapeutics
Ltd. Subsidiary), Clarity Pharmaceuticals Ltd., Bayer AG, Orano Med SAS, Isotopia Molecular Imaging Ltd., ITM Isotope
Technologies Munich SE, Johnson & Johnson, AdvanCell Isotopes Pty Ltd., Alpha-9 Theranostics Inc., Cancer Targeted
Technology LLC, FutureChem Co Ltd., Beijing Sinotau Intl. Pharmaceutical Technology Co., Ltd., Norroy Biosciences Co.
Ltd., RadioPharm Theranostics Limited, Precision Molecular, Inc., CellBion Co., Ltd., StarPharma Holdings Limited, Amgen
Inc., Crescendo Biologics Limited, Poseida Therapeutics, Inc., Regeneron Pharmaceuticals Inc., BioXcel Therapeutics,
Inc., Lava Therapeutics NV, Janux Therapeutics, Inc., Vir Therapeutics, Inc., Bivision Pharmaceuticals, Inc., GlyTherix Ltd,
Jiangsu Hengrui Pharmaceuticals Co., Ltd., and Full-Life Technologies Limited. Our competitors also include companies
developing other modalities to treat patients in metastatic castration-resistant prostate cancer ("mCRPC").
For TLX400-Tx (177Lu-DOTAGA.Glu.(FAPi)2), our main competitors are companies developing FAP-targeting
radiotherapeutics or diagnostics, including Novartis AG, Eli Lilly and Company Ltd, 3BP Pharmaceuticals GmbH, Ratio
Therapeutics Inc., Akiram Therapeutics, BoomRay Pharmaceuticals Co., Ltd., Philogen SPA, Precirix NV, Ratio
Therapeutics Inc., Spago Nanomedical AB, Yantai LNC, ITM Isotope Technologies Munich SE, Lantheus Holdings, Inc,
Perspective Therapeutics, Inc., Precision Molecular Inc., Sofie Biosciences, Inc., and GE Healthcare.
For TLX300-Tx (-olaratumab), our main competitors include companies with licensed soft-tissue sarcoma treatments
including Novartis AG, Adaptimmune Therapeutics plc, Agilent Technologies Inc., Boehringer Ingelheim GmbH, Esai Co.,
Ltd., , Mark, PharmaMar SA, Johnson & Johnson, Taiho Pharmaceuticals as well as other companies commercializing
chemotherapy regimens approved in soft-tissue sarcoma. Our competitors also include companies developing therapies
in the field of sarcoma including Ratio Therapeutics Inc., PTC Therapeutics, Polaris Pharmaceuticals, BioAtla. Eli Lilly and
Company Ltd, Advenchen Laboratories LLC, Intensity Therapeutics Inc, Sun Pharmaceutical Industries Ltd, Exelixis Inc,
Y-mAbs Therapeutics Inc, QBiotics Group Limited, Apollomics Inc, Pyxis Oncology, AADi Bioscience Inc, Moleculin
Biotech Inc, Cornerstone Pharmaceuticals Inc, Shasqi Inc, Noxopharm Limited, NANO MRNA Co Ltd, Avacta Group plc,
Iovance Biotherapeutics Inc, Foghorn Therapeutics Inc., OncoTherapy Science Inc. and Syena.
For TLX250-Tx (177Lu-DOTA-girentuximab), our main competitors include ITM Isotope Technologies Munich SE, Precision
Molecular, Inc., Norroy Biosciences Co. Ltd., PeptiDream Inc, Bristol Myers Squibb Company and Bayer AG. Our
competitors will also include companies developing other modalities to image renal cell carcinoma and carbonic
anhydrase IX.
For TLX101-Tx (iodofalan 131I), our main competitors include ITM Isotope Technologies Munich SE, Molecular Targeting
Technologies, Inc. ("MTTI"), Novartis AG, Radiopharm Theranostics Limited, Plus Therapeutics, Inc., Ariceum
Therapeutics GmbH, Boston Scientific Corporation, and Cellectar Biosciences, Inc. Our competitors will also include
companies developing other modalities to treat brain cancer.
For TLX090-Tx (153Sm-DOTMP), our main competitors include commercially available compounds that relieve pain from
osteoblastic bone metastases, including Novartis AG, Bayer AG, Pfizer Inc., Purdue Pharma L.P., Roxane Laboratories,
Inc., Johnson & Johnson, Mallinckrodt Inc., Endo Pharmaceuticals Holdings Inc., Mylan Laboratories Inc., Noven
Pharmaceuticals, Inc., Aveva Group PLC, Sandoz Group AG, Ranbaxy Laboratories Ltd., Amneal Pharmaceuticals LLC,
Hoffmann-La Roche AG, Apotex Inc, Orchid Healthcare LTD, Grünenthal GmbH, as well as other companies
commercializing or developing other palliative agents for osteoblastic bone metastases.
We are currently focused on developing and commercializing our Commercial Products and our product candidates for
the diagnosis and treatment of cancer and there are a variety of commercially available imaging and therapeutic
products marketed for cancer. In many cases, cancer imaging products and therapeutics are administered in combination
to enhance efficacy. Some of these products are branded and subject to patent protection, and others are available on a
generic basis or prepared under the practice of pharmacy or pharmacy compounding exemptions in certain jurisdictions.
Many of these products are well-established and are widely accepted by physicians, patients and third-party payors.
Insurers and other third-party payors may also encourage the use of generic diagnostics and therapeutics. Our
Commercial Products are, and any candidate for which we obtain marketing authorization will likely be, priced at a
22
significant premium over competitive generic products, also known as “home-brew” or “compounded” non-cGMP
products, which may make it difficult for us to achieve our business strategy of using our products in combination with
existing products or replacing existing products with our products, particularly if clinical differentiation or innovation
contribution is more limited compared to currently available products.
Further, our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize
products that are or are perceived to be more effective, safer, more tolerable, more convenient and/or less costly than
any of our currently approved products or product candidates which receive marketing authorization or that would
render our products obsolete or non-competitive. Our competitors may also obtain regulatory approval from the FDA or
other regulatory authorities for their product candidates more rapidly than we may obtain approval for ours, which could
result in our competitors establishing a stronger market position before we are able to enter the market or preventing us
from entering into a particular indication at all.
Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being
concentrated among a smaller number of our competitors. Smaller and other early-stage companies may also prove to
be significant competitors, particularly through collaborative arrangements with large and established companies. These
third parties compete with us in recruiting and retaining qualified scientific and management personnel, engaging clinical
trial sites and enrolling patients in clinical trials, as well as in acquiring technologies complementary to, or that may be
necessary for, our programs.
If we are not able to compete effectively against current or potential competitors, our business may be materially harmed
and our financial condition and results of operations will be adversely affected.
We may fail to achieve the degree of market acceptance by physicians, patients, third-party payors and others in the
medical community necessary for commercial success, of our Commercial Products and any product candidates for
which we obtain regulatory approval, including Illuccix and Gozellix, in which case we may not generate significant
revenues or remain profitable.
We may fail to gain sufficient market acceptance by physicians, patients, third-party payors and others in the medical
community necessary for commercial success of our Commercial Products and any product candidates for which we
obtain regulatory approval. Oncologists may be reluctant to switch their patients from existing therapies even when new
and potentially more effective or convenient treatments enter the market. Further, patients often acclimate to the
therapy that they are currently taking and do not want to switch unless their oncologists recommend switching products
or they are required to switch therapies due to lack of coverage and reimbursement for existing therapies.
Efforts to drive adoption within the medical community and third-party payors based on the benefits of our products and
product candidates require significant resources and may not be successful. The success of our Commercial Products
and our current or future product candidates, if approved, whether alone or in collaboration with third parties, including
achieving and maintaining an adequate level of market adoption, depends on several factors, including:
•our ability to successfully launch and achieve broad adoption of our Commercial Products or any other product for
which we obtain approval, or any future indications for which our Commercial Products may be approved;
•the competitive landscape for our Commercial Products and our product candidates, including the timing of new
competing products entering the market and the level and speed at which these products achieve market
acceptance;
•actual or perceived advantages or disadvantages of our Commercial Products or any product candidates for which
we obtain approval as compared to alternative treatments, including their respective safety, tolerability and efficacy
profiles, the potential convenience and ease of administration, access or cost effectiveness;
•the effectiveness of our sales, marketing, manufacturing and distribution strategies and operations;
•the consistency of any new data we collect and analyses we conduct with prior results; whether they support a
favorable safety, efficacy and effectiveness profile of our Commercial Products; and any potential impact on our
FDA or any foreign regulatory approvals and/or labeling for our Commercial Products;
•our ability to comply with the FDA’s and comparable foreign regulatory authorities’ post-marketing requirements
and commitments, including through successfully conducting, on a timely basis, additional studies that confirm
clinical efficacy, effectiveness and safety of our Commercial Products (or any product candidates for which we
obtain approval and are required to conduct such studies) and acceptance of the same by the FDA or similar foreign
regulatory authorities;
•acceptance of current indications of our Commercial Products and future indications of our Commercial Products
and other product candidates, if approved, by patients, the medical community and third-party payors;
•obtaining and maintaining coverage, adequate pricing and reimbursement by third-party payors, including
government payors, for our Commercial Products and our product candidates, if approved;
23
•the willingness of patients to pay out-of-pocket in the absence of third-party coverage or as co-pay amounts under
third-party coverage;
•our ability to enforce intellectual property rights in and to our products to prohibit a third-party from marketing a
competing product and our ability to avoid third-party post-grant patent proceeding or intellectual property
infringement claims;
•current and future restrictions or limitations on our approved or future indications and patient populations or other
adverse regulatory actions;
•the performance of our manufacturers, license partners, distributors, providers and other business partners, over
which we have limited control;
•any significant mis-estimations of the size of the market and market potential for any of our Commercial Products or
our product candidates;
•establishing and maintaining commercial manufacturing capabilities or making arrangements with third-party
manufacturers;
•the willingness of the target patient population to try new therapies and of physicians to prescribe these therapies,
based, in part, on their perception of our clinical trial data and/or the actual or perceived safety, tolerability and
effectiveness profile;
•maintaining an acceptable safety and tolerability profile of our Commercial Products or any of our product
candidates for which we obtain approval, including the prevalence and severity of any adverse events;
•the ability to offer our Commercial Products or any product candidates for which we obtain approval for sale at
competitive prices;
•adverse publicity about our Commercial Products or favorable publicity about competitive products; and
•our ability to maintain compliance with existing and new health care laws and regulations, including government
pricing, price reporting and other disclosure requirements related to such laws and regulations, and the potential
impact of such laws and regulations on physician prescribing practices and payor coverage.
If we do not achieve one or more of these factors in a timely manner, or at all, we could experience significant delays or
an inability to successfully commercialize our Commercial Products or our product candidates, if approved, which would
materially harm our business.
If we are unable to maintain or expand our sales, marketing and distribution capabilities, we may not be successful in
commercializing our Commercial Products or any of our product candidates, if approved.
We have built a commercial infrastructure in Australia, New Zealand, the U.S., Brazil, Canada and Europe for our
Commercial Products. Prior to building this infrastructure, we did not previously have any experience in the sales,
marketing or distribution of pharmaceutical products. If any of our product candidates are approved, we may need to
evolve our sales, marketing and distribution capabilities and we may not be able to do so successfully or on a timely
basis. In the future, we may choose to expand our sales, marketing and distribution infrastructure to market or co-
promote one or more of our product candidates, if and when they are approved, or enter into collaborations with respect
to the sale, marketing and distribution of our product candidates. We are working with existing and may in the future
work with additional partners to develop the commercial infrastructure to support the sale of our Commercial Products in
other jurisdictions.
There are risks involved with establishing and maintaining our own sales, marketing and distribution capabilities. For
example, recruiting and training a sales force is expensive and time-consuming and could delay any commercial launch
of a product candidate or negatively impact ongoing commercialization efforts for our approved products.
Further, we may underestimate the size of the sales force required for a successful product launch and we may need to
expand our sales force earlier and at a higher cost than we anticipated. If the commercial launch of any of our product
candidates is delayed or does not occur for any reason, including if we do not receive regulatory approval in the
timeframe we expect, we may have prematurely or unnecessarily incurred commercialization expenses. This may be
costly, and our investment would be lost if we cannot retain or reposition our sales and marketing personnel.
Factors that may inhibit our efforts to successfully commercialize our Commercial Products or any of our product
candidates, if approved, on our own include:
•our inability to recruit, train and retain adequate numbers of effective sales, market access, market analytics,
operations and marketing personnel;
•the inability of sales personnel to obtain access to physicians or persuade adequate numbers of physicians to
prescribe current or future products;
24
•the lack of complementary products, which may put us at a competitive disadvantage relative to companies with
more extensive product lines;
•unforeseen costs and expenses associated with creating an independent sales, marketing and distribution
organization;
•our inability to obtain sufficient coverage and reimbursement from third-party payors and governmental agencies;
•our ability to supply, manufacture and deliver sufficient inventory of our products for commercial sale on a timely
basis; and
•existing or new competitors taking share from our Commercial Products or any other product candidate for which
we obtain approval in the future, or preventing our Commercial Products or any such product from gaining share in
its approved indications.
The commercial success of our Commercial Products and our product candidates, if approved, will depend upon
public perception of radiopharmaceuticals and the degree of their market acceptance by physicians, key opinion
leaders, patients, healthcare payors and others in the medical community.
Adverse events in clinical trials of our product candidates, or in clinical trials or other studies conducted by others
involving similar products, which may include the same radioisotopes as our Commercial Products and/or our product
candidates, and the resulting negative publicity, as well as any other adverse events in the field of radiopharmaceuticals
that may occur in the future, could result in a decrease in demand for our Commercial Products or any future product
candidates that we may develop and for which we obtain regulatory approval. If public perception is influenced by claims
that radiopharmaceuticals or specific therapies within radiopharmaceuticals are unsafe, our Commercial Products or any
product candidates for which we obtain regulatory approval may not be accepted by the general public or the medical
community.
In particular, the commercial success of our Commercial Products and our product candidates, if approved, will depend
upon, among other things, these products gaining and maintaining acceptance by physicians, key opinion leaders,
patients, third-party payors, and other members of the medical community as efficacious and cost-effective alternatives
to competing products and treatments. If our Commercial Products or any of our product candidates, once approved, do
not achieve and maintain an adequate level of acceptance, we may not generate material sales of that product or be able
to successfully commercialize it. The degree of market acceptance of our Commercial Products or our product
candidates, if approved, will depend on a number of factors, including:
•our ability to provide acceptable evidence of safety and efficacy;
•the prevalence and severity of any adverse events in general, and differentiation relative to other treatments;
•limitations or warnings contained in the labeling approved by the FDA;
•the size of the target patient population;
•advertising concerning our products or competing products and treatments;
•availability, relative cost and relative efficacy of alternative and competing treatments;
•the ability to offer our products for sale at competitive prices;
•the relative convenience and ease of administration of our Commercial Products and product candidates, if
approved, which may require coordination amongst multiple physicians across disciplines for administration;
•the willingness of the target patient population to try new products and of physicians to prescribe these products;
•strength of marketing and distribution support;
•publicity for our Commercial Products and competing products and treatments;
•the existence of distribution and/or use restrictions, such as through a REMS;
•the availability of third-party payor coverage and adequate reimbursement;
•the timing of any marketing approval in relation to other product approvals;
•support from patient advocacy groups;
•any restrictions on the use of our products together with other medications; and
25
•the sufficiency of coverage or reimbursement by third parties.
Manufacturing of radiopharmaceuticals is complex and we may encounter difficulties in production. If we encounter
such difficulties, our ability to provide supply of our Commercial Products or any of our product candidates for
preclinical studies and clinical trials or for commercial purposes could be delayed or stopped.
Manufacturing of radiopharmaceuticals is complex, highly regulated and must comply with cGMPs and similar foreign
requirements. While we have manufacturing capabilities of our own, we also rely on third parties, such as contract
manufacturing organizations ("CMOs"), for the manufacture of our Commercial Products and our product candidates. If
we are unable to obtain or maintain arrangements with CMOs, or to do so on commercially reasonable terms, we may not
be able to commercialize our Commercial Products or develop our product candidates successfully. Our third-party
manufacturing providers may not be able to provide adequate resources or capacity to meet our needs on a timely basis
or at all, and may incorporate their own proprietary processes into our product candidate manufacturing processes. We
have limited control and oversight of a third-party’s proprietary process, and a third-party may elect to modify its
process without our consent or knowledge. These modifications could negatively impact our manufacturing, including
product loss or failure that requires additional manufacturing runs or a change in manufacturer, either of which could
significantly increase the cost of and significantly delay the manufacture of our Commercial Products or any of our
product candidates.
Additionally, as we expect the market for our Commercial Products and PSMA-PET imaging to expand and our product
candidates to progress through preclinical studies and clinical trials towards potential approval and commercialization, it
is possible that various aspects of manufacturing will be altered in an effort to optimize processes and results. Such
changes may require new submissions to and approval from regulators, which may further delay the timeframes under
which modified manufacturing processes can be used for our Commercial Products or any of our product candidates,
and additional bridging studies or trials may be required. Any such delay could harm our business, financial condition,
results of operations and prospects.
We, our contract manufacturers, any future collaborators and their contract manufacturers could be subject to periodic
unannounced inspections by the FDA or other comparable foreign regulatory authorities, to monitor and ensure
compliance with cGMPs or similar foreign requirements. Despite our efforts to audit and verify regulatory compliance, we
or one or more of our third-party manufacturing vendors may be found on regulatory inspection by the FDA or other
comparable foreign regulatory authorities to be noncompliant with cGMPs or similar foreign regulations. This may result
in shutdown of our facility or that of the third-party vendor or invalidation of product lots or processes, which could
adversely affect our business, financial condition, results of operations and prospects. In some cases, a product recall
may be warranted or required, which would materially affect our ability to supply and market our products and could be
costly and result in reputational damage.
We may be unable to generate and/or obtain a sufficient supply of radioisotopes to support clinical development or
manufacturing at commercial scale.
As a radiopharmaceutical company, our Commercial Products and our product candidates are prepared for patient
administration using radioisotopes. Gallium-68 (68Ga) is a necessary component isotope for radiopharmacies to radiolabel
our Commercial Products for patient administration and is sourced by a radiopharmacy directly. Other important isotopes
applicable to our current pipeline of diagnostic and therapeutic product candidates include zirconium-89 (89Zr),
lutetium-177 (177Lu), yttrium-90 (90Y), fluorine-18 (18F), iodine-131 (131I), technetium-99m or (99mTc), actinium-225 (225Ac),
astatine-211 (211At), and Samarium-153 (153Sm). We procure these isotopes from suppliers based predominantly in
Canada or Europe. Global isotope supply chains, including obtaining precursor or raw materials necessary to produce
many of the synthetic radioisotopes used in nuclear medicine, are commonly sourced from countries such as Russia,
Brazil, South Africa and Türkiye that may, from time-to-time, be subject to instability, unrest, protests, intergovernmental
conflicts and various international trade or monetary sanctions. Where isotopes or raw materials are procured under
various medical or humanitarian exemptions, including countries that may, from time-to-time, be subject to instability,
unrest, protests, intergovernmental conflicts and various international trade or monetary sanctions, those exemptions
may be repealed or altered in a way that is detrimental to our ability to operate our business.
We aim to maintain multiple supply agreements with isotope suppliers and stockpiles to ensure adequate quantities to
meet our current pipeline development needs. However, there is a limited supply of some radioisotopes due to the
limited supply of starting radioactive raw materials to create the radioisotope or the complexity required to manufacture
isotopes to the required quality and purity standards for effective radiolabeling. We aim to maintain supply relationships
with all major current suppliers and for certain isotopes there are no or limited alternatives to our current suppliers.
While we are making investments to secure additional access to and capabilities for manufacturing isotopes, we may
encounter supply shortages which could affect our business operations and results of operations. There can be no
assurance that our suppliers will renew existing contracts on acceptable terms, or even at all. Additionally, failure to
acquire enough medical-grade isotopes for specific product candidates would make it impossible to effectively complete
clinical trials, especially as we scale up for later-stage clinical trials, and to commercialize any product candidates that
we may develop, which would materially harm our business.
Isotope suppliers may also have limited production capacity to meet future commercial demand, and there is no
guarantee that production will start in the time frame we expect. Even where a contract exists, we may have limited
recourse if a supplier is unable to meet its obligations. Suppliers may also be unable to meet their obligations for any
26
number of reasons. For example, the U.S. Department of Energy has reserved its ability to cancel private orders when the
supply is instead needed for national defense, environmental safety, or in the event of any other sort of lack of supply
capacity or for a number of other reasons that are outside of our control.
Radioisotopes or radioactive raw materials may only be available from a limited number of countries, including Russia,
Brazil, Türkiye or South Africa. Our isotope suppliers obtain the radioactive materials from source material countries in
accordance with applicable laws and export regulations, usually under medical exemption, and then use the raw
materials to manufacture the radioisotopes for onward clinical sale and commercial sale to third parties, including
governments, hospitals and pharmaceutical companies. We and our suppliers are exposed to a number of environmental
and geopolitical risks beyond radioactive raw material availability, including restrictions on trade of certain items with
Russia, and other unforeseen geopolitical factors that limit our ability to access our supply of raw material. The ongoing
war in Ukraine and subsequent economic sanctions imposed on Russia, including by the U.S., may impact our ability to
procure supply of necessary isotopes and may impact our product development timelines. For example, while our current
suppliers are not currently designated on any export or sanctions-related restricted party lists maintained by the U.S.
government, there is no guarantee our suppliers (or their third-party suppliers of raw materials) will not be designated on
such lists in the future. In addition, our dependence on international radioisotope suppliers is increased in the near term
because the U.S. Department of Energy restricts usage for certain isotopes for clinical development outside the U.S., and
therefore, we must rely on our suppliers for our international operations. To date, the ongoing war in Ukraine has not
materially impacted the development of any of our product candidates, nor has it materially impacted the price at which
we are able to purchase isotopes. Although we do not expect to encounter additional delays from our suppliers based on
the ongoing war in the Ukraine, we may experience delays in the future, and any such delay could have an adverse
material impact on our development plans and business. We expect to continue to monitor and adapt our development
plans as necessary in response to environmental and geopolitical risks. Any difficulty that our suppliers have in procuring
raw materials may also magnify the impact of other risks described in this Annual Report.
Our ability to conduct clinical trials to advance our product candidates is dependent on our ability to either self-generate
and/or obtain these radioisotopes and other isotopes we may choose to utilize in the future. While we intend to scale-up
our manufacturing facilities to achieve vertical integration and the ability to self-manufacture our final diagnostics and
therapeutics products, we are dependent on third-party manufacturers and suppliers for many of our isotopes, and our
suppliers will be dependent on third parties to supply the raw radioactive materials. These parties may not perform their
contracted services or may breach or terminate their agreements with us. Our suppliers are subject to regulations and
standards that are overseen by regulatory and government agencies, and we have no control over our suppliers’
compliance with these standards. Failure to comply with regulations and standards may result in their inability to supply
an isotope that could result in delays in our clinical trials or commercialization, which could have a negative impact on our
business.
Even if we are able to effectively commercialize our Commercial Products or any product candidates for which we
obtain approval, the products may not receive coverage or may become subject to unfavorable pricing regulations,
third-party reimbursement practices or healthcare reform initiatives, all of which would harm our business.
The legislation and regulations that govern regulatory approvals, pricing, coverage and reimbursement for new imaging
and therapy products vary widely from country to country. As a result, we might obtain regulatory approval for a product
in a particular country, but then be subject to pricing or reimbursement regulations that delay the commercial launch of
the product, possibly for lengthy time periods, and negatively impact the revenues we are able to generate from product
sales in that country. In the U.S. and most other major markets internationally, approval and reimbursement decisions are
not linked directly, but there is increasing scrutiny from the Congress, government or regulatory authorities, payors,
patient organizations of the pricing or reimbursement of pharmaceutical products. Adverse pricing or reimbursement
limitations may also hinder our ability to recoup our investment in one or more product candidates, even if our product
candidates obtain regulatory approval.
Our ability to successfully commercialize our Commercial Products and any other products that we may develop or
acquire will depend, in part, on the extent to which satisfactory pricing, coverage and reimbursement for these products
is available from government payors, private health insurers and other organizations. Government authorities and third-
party payors, such as private health insurers and health maintenance organizations, decide which medications they will
pay for and establish reimbursement levels. Obtaining and maintaining adequate coverage and reimbursement for our
Commercial Products and any of our product candidates, if approved, may be difficult. Moreover, the process for
determining whether a third-party payor will provide coverage for a product may be separate from the process for
setting the price of a product or for establishing the reimbursement rate that such a payor will pay for the product.
Further, one payor’s determination to provide coverage for a product does not assure that other payors will also provide
coverage and reimbursement for our products. Even with payor coverage, patients may be unwilling or unable to pay the
copay required and may choose not to take or use our products.
A primary trend in the healthcare industry in the U.S. and elsewhere is cost containment. Government authorities and
third-party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular
medications. Increasingly, third-party payors are requiring that drug companies provide them with predetermined
discounts from list prices and are challenging the prices charged for medical products.
Third-party payors may also seek, with respect to an approved product, additional clinical evidence that goes beyond
the data required to obtain regulatory approval. They may require such evidence to demonstrate clinical benefits and
value in specific patient populations or they may call for costly pharmaceutical studies to justify coverage and
27
reimbursement or the level of reimbursement relative to other therapies before covering our products. Accordingly, we
cannot be sure that reimbursement will be or will continue to be available for our Commercial Products and any product
that we commercialize in the future and, if reimbursement is available, we cannot be sure as to the level of
reimbursement and whether it will be adequate. Coverage and reimbursement may impact the demand for or the price of
our Commercial Products or any product candidate for which we obtain regulatory approval. If reimbursement is not
available or is available only at limited levels, we may not be able to successfully commercialize our Commercial Products
or any other approved products.
There may be significant delays in obtaining reimbursement for newly approved products, and coverage may be more
limited than the indications for which the product is approved by the FDA or comparable foreign regulatory authorities.
Moreover, eligibility for reimbursement does not imply that our Commercial Products or any product candidate for which
we obtain approval will be paid for in all cases or at a rate that covers our costs, including research, development,
manufacture, sale and distribution. Interim reimbursement levels for new products, if applicable, may also not be
sufficient to cover our costs and may not be made permanent. Reimbursement rates may vary according to the use of
the product and the clinical setting in which it is used, may be based on reimbursement levels already set for lower cost
products and may be incorporated into existing payments for other services. Net prices for products may be reduced by
mandatory discounts or rebates required by government healthcare programs or private payors and by any future
relaxation of laws that presently restrict imports of products from countries where they may be sold at lower prices than
in the U.S. Third-party payors in the U.S. often rely upon Medicare coverage policy and payment limitations in setting
their own reimbursement policies. Our inability to promptly obtain coverage and profitable payment rates from both
government-funded and private payors for any approved products that we develop could have a material adverse effect
on our operating results, our ability to raise capital needed to commercialize our products and our overall financial
condition.
Product liability lawsuits against us could divert our resources, cause us to incur substantial liabilities and limit
commercialization of our Commercial Products or any other products or product candidates that we may develop or
acquire.
We face an inherent risk of product liability exposure related to our commercialization of our Commercial Products and
the testing of our product candidates in human clinical trials as the administration of our products to humans may expose
us to liability claims, whether or not our products are actually at fault for causing any harm or injury. As our Commercial
Products are used over longer periods of time by a wider group of patients taking numerous other medicines or by
patients with additional underlying conditions, the likelihood of adverse product reactions or unintended adverse events,
including death, may increase. For example, we may be sued if any of our Commercial Products or product candidates
we develop allegedly causes injury or is found to be otherwise unsuitable during clinical testing, manufacturing,
marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in
design, a failure to warn of dangers inherent in the product, negligence, strict liability or a breach of warranties. Claims
could also be asserted under state consumer protection acts. If we cannot successfully defend ourselves against claims
that our products or product candidates caused injuries, we will incur substantial liabilities or may be required to limit
commercialization of our Commercial Products and product candidates, if approved. Regardless of merit or eventual
outcome, liability claims may result in:
•decreased demand for our Commercial Products and any other products that we may develop or acquire;
•injury to our reputation and significant negative media attention;
•loss of critical partners and/or partnership agreements;
•withdrawal of clinical trial participants;
•initiation of investigations by regulators;
•product recalls, withdrawals or labeling, marketing or promotional restrictions;
•significant costs to defend the related litigation;
•substantial monetary awards to trial participants or patients;
•loss of revenue;
•reduced resources of our management to pursue our business strategy; and
•the inability to successfully commercialize our Commercial Products and any other products that we may develop or
acquire.
We currently hold clinical trial liability insurance of up to US$20 million per occurrence in the aggregate and general
product liability insurance coverage in the amount of US$20 million in the aggregate, but that coverage may not be
adequate to cover any and all liabilities that we may incur. Insurance coverage is increasingly expensive. We may not be
able to maintain insurance coverage at a reasonable cost or in an amount adequate to satisfy any liability that may arise.
28
We may be subject to securities class action litigation or other shareholder litigation, which could result in
substantial costs and divert management's attention and resources.
Companies that have experienced volatility in the market price of their securities have frequently been subject to
securities class action litigation or other shareholder derivative actions. We may become the target of this type of
litigation in the future, particularly if we experience significant stock price volatility or declines, announce material
restatements of financial results, fail to meet earnings expectations, or disclose material weaknesses in our internal
controls, such as we have disclosed in our management report on internal control over financial reporting. Additionally,
public companies are increasingly subject to litigation related to corporate governance matters, executive compensation,
environmental, social and governance ("ESG") disclosures, and other matters that may give rise to shareholder claims or
derivative actions.
Securities litigation and shareholder derivative actions, regardless of their outcome or merit, can result in substantial
costs, including significant legal fees and expenses, damages, and settlement amounts. Such litigation may also divert
management's attention and resources away from business operations, which could adversely affect our ability to
execute on our strategic initiatives. The defense or settlement of any such litigation could have a material adverse effect
on our business, financial condition, results of operations, and cash flows.
Furthermore, any adverse judgment or settlement in connection with shareholder litigation could result in negative
publicity and reputational harm, which may further impact our stock price and ability to attract and retain key personnel.
The outcome of complex legal proceedings is inherently unpredictable and subject to significant uncertainties, and we
may not be able to accurately estimate potential liabilities associated with any pending or threatened litigation.
Our directors' and officers' liability insurance may not cover all potential claims or may not be adequate to indemnify us
for all liability that may be imposed. Any future claims could also make it more difficult and expensive to obtain adequate
insurance coverage
Risks Related to Regulatory Matters
Even if we complete the necessary preclinical studies and clinical trials for our product candidates, the regulatory
approval process is expensive, time-consuming and uncertain and we or they may not receive approvals for the
commercialization of some or all of our product candidates in a timely manner, or at all.
Our long-term success and ability to sustain and grow revenue depends on our ability to continue to successfully
develop our product candidates and obtain regulatory approval to market our products both in and outside of the U.S. In
order to market and sell our products in the European Union and many other jurisdictions, we must obtain separate
marketing approvals and comply with numerous and varying regulatory requirements. The FDA and comparable foreign
regulatory authorities, whose laws and regulations may differ from country to country, impose substantial requirements
on the development of product candidates to become eligible for marketing approval, have substantial discretion in the
process, and may refuse to accept any application or may decide that the data are insufficient for approval and require
additional preclinical studies, clinical trials or other studies and testing. The time required to obtain approval outside of
the U.S. may differ substantially from that required to obtain FDA approval. For example, in many countries outside of the
U.S., it is required that the drug also be approved for reimbursement before the drug can be sold in that country.
Approval by the FDA does not ensure approval by regulatory authorities in other countries or jurisdictions, and approval
by one regulatory authority outside of the U.S. does not ensure approval by regulatory authorities in other countries or
jurisdictions or by the FDA. However, a failure or delay in obtaining regulatory approval in one country may have a
negative effect on the regulatory process in other countries.
In addition, the FDA and foreign regulatory authorities retain broad discretion in evaluating the results of our clinical trials
and in determining whether the results demonstrate that any product candidate is safe and effective. If we are required
to conduct additional clinical trials of our Commercial Products prior to approval of any additional investigational
indications we are developing them for, or of any product candidates prior to approval, we may need substantial
additional funds, and there is no assurance that the results of any such additional clinical trials will be sufficient for
approval.
The process of obtaining marketing approvals, both in the U.S. and abroad, is lengthy, expensive and uncertain. It may
take many years, if approval is obtained at all, and can vary substantially based upon a variety of factors, including the
type, complexity and novelty of the product candidates involved. Securing marketing approval requires the submission of
extensive preclinical and clinical data and supporting information, including manufacturing information, to regulatory
authorities for each indication to establish the product candidate’s safety and efficacy.
In addition, changes in or the enactment of additional statutes, promulgation of regulations or issuance of guidance
during preclinical or clinical development, or comparable changes in the regulatory review process for each submitted
product application, may cause delays in the approval or rejection of an application. For example, in December 2022,
with the passage of Food and Drug Omnibus Reform Act ("FDORA"), Congress required sponsors to develop and submit
a Diversity Action Plan ("DAP"), for each Phase 3 clinical trial or any other “pivotal study” of a new drug or biological
product. These plans are meant to encourage the enrollment of more diverse patient populations in late-stage clinical
trials of FDA regulated products. In June 2024, as mandated by FDORA, the FDA issued draft guidance outlining the
general requirements for DAPs. Unlike most guidance documents issued by the FDA, the DAP guidance when finalized
29
will have the force of law because FDORA specifically dictates that the form and manner for submission of DAPs are
specified in FDA guidance. On January 27, 2025, in response to an Executive Order issued by President Trump on
January 21, 2025, on Diversity, Equity and Inclusion programs, the FDA removed this draft guidance from its website.
This action raises questions about the applicability of statutory obligations to submit DAPs and the agency’s current
thinking on best practices for clinical development.
Further, on January 31, 2022, the new Clinical Trials Regulation (EU) No 536/2014 became applicable in the European
Union and replaced the prior Clinical Trials Directive 2001/20/EC. The new regulation aims at simplifying and streamlining
the authorization, conduct and transparency of clinical trials in the European Union. Under the new coordinated
procedure for the approval of clinical trials, the sponsor of a clinical trial to be conducted in more than one EU Member
State will only be required to submit a single application for approval. The submission will be made through the Clinical
Trials Information System, a new clinical trials portal overseen by the European Medicines Agency ("EMA"), and available
to clinical trial sponsors, competent authorities of the EU Member States and the public. We have not previously secured
authorization to conduct clinical studies in the European Union pursuant to this new regulation and, accordingly, there is
a risk that we may be delayed in commencing such studies.
The FDA or other regulatory authorities may determine that (i) our product candidates are not safe and effective, are only
moderately effective or have undesirable or unintended adverse events, toxicities or other characteristics that preclude
our obtaining marketing approval or prevent or limit commercial use; (ii) the dose used in a clinical trial has not been
optimized and require us to conduct additional dose optimization studies; or (iii) the comparator arm in a trial is no longer
the appropriate comparator due to the evolution of the competitive landscape or subsequent data of the comparator
product, even if the FDA or other regulatory authority had previously approved the trial design, and we may be required
to amend the trial or we may not receive approval of the indication.
Moreover, principal investigators for our future clinical trials may serve as scientific advisors or consultants to us and
receive compensation in connection with such services. Under certain circumstances, we may be required to report
some of these relationships to the FDA or comparable foreign regulatory authorities. The FDA or a comparable foreign
regulatory authority may conclude that a financial relationship between us and a principal investigator has created a
conflict of interest or otherwise affected interpretation of the study. The FDA or a comparable foreign regulatory
authority may therefore question the integrity of the data generated at the applicable clinical trial site and the utility of
the clinical trial itself may be jeopardized. This could result in a delay in approval, or rejection, of our marketing
applications by the FDA or a comparable foreign regulatory authority, as the case may be, and may ultimately lead to the
denial of marketing approval of one or more of our product candidates.
Further, under the Pediatric Research Equity Act ("PREA"), an NDA, BLA or supplement to an NDA or BLA for certain
drugs and biological products must contain data to assess the safety and effectiveness of the drug or biological product
in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for
which the product is safe and effective, unless the sponsor receives a deferral or waiver from the FDA. A deferral may be
granted for several reasons, including a finding that the product or therapeutic candidate is ready for approval for use in
adults before pediatric trials are complete or that additional safety or effectiveness data needs to be collected before the
pediatric trials begin. The applicable legislation in the European Union also requires sponsors to either conduct clinical
trials in a pediatric population in accordance with a Pediatric Investigation Plan approved by the Pediatric Committee of
the EMA, or to obtain a waiver or deferral from the conduct of these studies by this Committee. For any of our product
candidates for which we are seeking regulatory approval in the U.S. or the European Union, we cannot guarantee that we
will be able to obtain a waiver or alternatively complete any required studies and other requirements in a timely manner,
or at all, which could result in associated reputational harm and subject us to enforcement action.
In addition, we could be adversely affected by several significant administrative law cases decided by the U.S. Supreme
Court in 2024. In Loper Bright Enterprises v. Raimondo, for example, the court overruled Chevron U.S.A., Inc. v. Natural
Resources Defense Council, Inc., which for 40 years required federal courts to defer to permissible agency
interpretations of statutes that are silent or ambiguous on a particular topic. The U.S. Supreme Court stripped federal
agencies of this presumptive deference and held that courts must exercise their independent judgment when deciding
whether an agency such as the FDA acted within its statutory authority under the Administrative Procedure Act ("APA").
Additionally, in Corner Post, Inc. v. Board of Governors of the Federal Reserve System, the court held that actions to
challenge a federal regulation under the APA can be initiated within six years of the date of injury to the plaintiff, rather
than the date the rule is finalized. The decision appears to give prospective plaintiffs a personal statute of limitations to
challenge longstanding agency regulations. Another decision, Securities and Exchange Commission v. Jarkesy,
overturned regulatory agencies’ ability to impose civil penalties in administrative proceedings. These decisions could
introduce additional uncertainty into the regulatory process and may result in additional legal challenges to actions taken
by federal regulatory agencies, including the FDA and Centers for Medicare & Medicaid Services ("CMS"), that we rely
on. In addition to potential changes to regulations as a result of legal challenges, these decisions may result in increased
regulatory uncertainty and delays and other impacts, any of which could adversely impact our business and operations.
Finally, with the change in presidential administrations in 2025, there is substantial uncertainty as to how, reductions in
force, changes in leadership, and modifications or revisions to the requirements and policies of the FDA and other
regulatory agencies with jurisdiction over our product candidates will impact our business and operations. The
uncertainty could present new challenges or potential opportunities as we navigate the clinical development and
approval process for our product candidates.
30
The approval of our product candidates for commercial sale could also be delayed, limited or denied or we may be
required to conduct additional studies for a number of reasons, including, but not limited to, the following:
•regulatory authorities may determine that our product candidates do not demonstrate safety and effectiveness in
accordance with regulatory agency standards based on a number of considerations, including adverse events that
are reported during clinical trials;
•regulatory authorities could analyze and/or interpret data from clinical trials and preclinical testing in different ways
than we interpret them and determine that our data is insufficient for approval;
•regulatory authorities may require more information, including additional preclinical or clinical data or the conduct of
new trials, to support approval;
•regulatory authorities could determine that our manufacturing processes are not properly designed, are not
conducted in accordance with federal or other laws or otherwise not properly managed, and we may be unable to
obtain regulatory approval for a commercially viable manufacturing process for our product candidates in a timely
manner, or at all;
•the supply or quality of our product candidates for our clinical trials may be insufficient, inadequate or delayed;
•the size of the patient population required to establish the efficacy of our product candidates to the satisfaction of
regulatory agencies may be larger than we or they anticipated;
•our failure or the failure of clinical sites, and the records kept at the respective locations, including records
containing clinical trial data, to be in compliance with the FDA’s GCP, requirements or comparable regulations
outside of the U.S.;
•regulatory authorities may change their approval policies or adopt new regulations;
•regulatory authorities may not be able to undertake reviews of our marketing applications, conduct applicable
inspections or proceed through their approval processes in a timely manner;
•the results of our earlier clinical trials may not be representative of our future, larger trials;
•regulatory authorities may not agree with our regulatory approval strategies or components of our or their
regulatory filings, such as the design or implementation of the relevant clinical trials; or
•a product may not be approved for the indications that we request or may be limited or subject to restrictions or
post-approval commitments that render the approved drug not commercially viable.
Accordingly, we may not be able to submit applications for marketing approvals/authorizations and may not receive
necessary approvals to commercialize our products in any market. Any failure, delay or setback in obtaining regulatory
approval for our product candidates could materially adversely affect our ability to generate revenue from a particular
product candidate, which could result in significant harm to our financial position and adversely impact the price of our
ordinary shares and ADSs.
Failure to obtain marketing approval in foreign jurisdictions would prevent our medicines from being marketed in
such jurisdictions and any of our medicines that are approved for marketing in such jurisdiction will be subject to risk
associated with foreign operations.
In order to market and sell our medicines in the European Union and many other foreign jurisdictions, we or our
collaborators must obtain separate marketing approvals and comply with numerous and varying regulatory requirements.
The approval procedure varies among countries and can involve additional testing. The time required to obtain approval
may differ substantially from that required to obtain FDA approval. The regulatory approval process outside the U.S.
generally includes all of the risks associated with obtaining FDA approval. In addition, in many countries outside the U.S.,
a product must be approved for reimbursement before the product can be approved for sale in that country. We or our
collaborators may not obtain approvals from regulatory authorities outside the U.S. on a timely basis, if at all. Moreover,
approval by the FDA does not ensure approval by regulatory authorities in other countries or jurisdictions, and approval
by one regulatory authority outside the U.S. does not ensure approval by regulatory authorities in other countries or
jurisdictions or by the FDA.
Additionally, we could face heightened risks with respect to obtaining marketing authorization in the United Kingdom as a
result of the withdrawal of the United Kingdom from the European Union, commonly referred to as Brexit. The United
Kingdom is no longer part of the European Single Market and EU Customs Union. As of January 1, 2025, the Medicines
and Healthcare Products Regulatory Agency ("MHRA"), is responsible for approving all medicinal products destined for
the United Kingdom market (i.e., Great Britain and Northern Ireland). At the same time, a new international recognition
procedure ("IRP"), will apply, which intends to facilitate approval of pharmaceutical products in the United Kingdom. The
IRP is open to applicants that have already received an authorization for the same product from one of the MHRA’s
specified Reference Regulators ("RRs"). The RRs notably include EMA and regulators in the EU/European Economic Area
("EEA") member states for approvals in the EU centralized procedure and mutual recognition procedure as well as the
31
FDA (for product approvals granted in the U.S.). However, the concrete functioning of the IRP is currently unclear. Any
delay in obtaining, or an inability to obtain, any marketing approvals may force us or our collaborators to restrict or delay
efforts to seek regulatory approval in the United Kingdom for our product candidates, which could significantly and
materially harm our business.
In addition, foreign regulatory authorities may change their approval policies and new regulations may be enacted. For
instance, the EU pharmaceutical legislation is currently undergoing a complete review process, in the context of the
Pharmaceutical Strategy for Europe initiative, launched by the European Commission in November 2020. The European
Commission’s proposal for revision of several legislative instruments related to medicinal products was published on April
26, 2023. In December 2025, the Council of the European Union and the European Parliament reached provisional
agreement on a comprehensive overhaul of the EU legislative framework for pharmaceuticals. There are a number of
significant changes, including a potential reduction in regulatory data protection, a streamlining of the regulatory
approval process potentially allowing products to reach the market in a shorter period of time, and introduction of
shortage prevention plans. The agreement still requires formal approval and adoption. If adopted, there will be a
transitional period (between 18-36 months) for implementation. The revisions may have a significant impact on the
pharmaceutical industry in general and our business in the long term.
We expect that we will be subject to additional risks in commercializing any of our product candidates that receive
marketing approval outside the U.S., including tariffs, trade barriers and regulatory requirements; economic weakness,
including inflation, or political instability in particular foreign economies and markets; compliance with tax, employment,
immigration and labor laws for employees living or traveling abroad; foreign currency fluctuations, which could result in
increased operating expenses and reduced revenue, and other obligations incident to doing business in another country;
and workforce uncertainty in countries where labor unrest is more common than in the U.S. In addition, we have limited
experience commercializing products outside of the U.S. and such efforts may depend on our ability to find a suitable
collaborator.
We intend to conduct certain of our clinical trials globally. However, the FDA and other foreign equivalents may not
accept data from such trials, in which case our development plans will be delayed, which could materially harm our
business.
We have conducted and intend to continue conducting certain of our clinical trials globally. The acceptance by the FDA
or other regulatory authorities of study data from clinical trials conducted outside their jurisdiction may be subject to
certain conditions or may not be accepted at all. In cases where data from foreign clinical trials are intended to serve as
the sole basis for marketing approval in the U.S., the FDA will generally not approve the application on the basis of
foreign data alone unless (i) the data are applicable to the U.S. population and U.S. medical practice; (ii) the trials were
performed by clinical investigators of recognized competence and pursuant to GCP regulations; and (iii) the data may be
considered valid without the need for an on-site inspection by the FDA, or if the FDA considers such inspection to be
necessary, the FDA is able to validate the data through an on-site inspection or other appropriate means.
In addition, even where the foreign study data are not intended to serve as the sole basis for approval, the FDA will not
accept the data as support for an application for marketing approval unless the study is well-designed and well-
conducted in accordance with GCP requirements and the FDA is able to validate the data from the study through an
onsite inspection if deemed necessary. Many foreign regulatory authorities have similar approval requirements. In
addition, such foreign trials would be subject to the applicable local laws of the foreign jurisdictions where the trials are
conducted. There can be no assurance that the FDA or any comparable foreign regulatory authority will accept data from
trials conducted outside of the U.S. or the applicable jurisdiction. If the FDA or any comparable foreign regulatory
authority does not accept such data, it would result in the need for additional trials, which could be costly and time-
consuming, and which may result in current or future product candidates that we may develop not receiving approval for
commercialization in the applicable jurisdiction.
Conducting clinical trials outside the U.S. also exposes us to additional risks, including risks associated with:
•additional foreign regulatory requirements;
•foreign exchange fluctuations;
•compliance with foreign manufacturing, customs, shipment and storage requirements;
•cultural differences in medical practice and clinical research;
•diminished protection of intellectual property in some countries; and
•interruptions or delays in our trials resulting from geopolitical events, such as tariffs, other trade restrictions, armed
conflict, or political instability.
Products utilizing our technology may need to be approved or cleared by the FDA and similar regulatory agencies or
certified by notified bodies worldwide as medical devices. We may not receive, or may be delayed in receiving, the
necessary approval, clearance or certification for our future medical device products, which would adversely affect
business, financial condition, results of operations and prospects.
32
We are developing artificial intelligence ("AI") and surgical assistance offerings that may be subject to regulation as
medical devices in the U.S. and other jurisdictions. We have not yet utilized our AI platform in the development of our
Commercial Products or product candidates. To date, we have not had any discussion with the FDA or other regulatory
authorities or notified bodies regarding the regulatory pathways required to market these technologies. The FDA or
similar regulatory agencies may subject these offerings to medical device requirements, including premarket review,
lengthier or more rigorous processes than we expected that may include the performance of one or more clinical trials.
Efforts to achieve requisite governmental clearances and approvals could be costly and time consuming, and we may not
be able to obtain any such required clearances or approvals in accordance with our anticipated timeline or in a cost-
efficient manner. Any delay or failure to obtain necessary regulatory clearances, approvals or certifications could have a
material negative impact on our ability to generate revenues.
The FDA categorizes medical devices into one of three classes - Class I, II, or III - based on the risks presented by the
device and the regulatory controls necessary to provide a reasonable assurance of the device’s safety and effectiveness.
Class I includes devices with the lowest risk to the patient and are those for which safety and effectiveness can be
assured by adherence to the FDA’s General Controls for medical devices, which include compliance with the applicable
portions of the Quality System Regulation ("QSR") facility registration and product listing, reporting of adverse medical
events or certain malfunctions, and truthful and non-misleading labeling, advertising, and promotional materials. Class II
devices are subject to the FDA’s General Controls, and special controls as deemed necessary by the FDA to ensure the
safety and effectiveness of the device. Special controls are established by the FDA for a specific device type and often
include specific labeling provisions, performance metrics, and other types of controls that mitigate risks of the device.
Devices deemed by the FDA to pose the greatest risks, such as life-sustaining, life-supporting or some implantable
devices, or devices that have a new intended use, or use advanced technology that is not substantially equivalent to that
of a legally marketed device, are placed in Class III, requiring approval of a premarket approval application ("PMA").
In the U.S., before we can market a new medical device, or a new use of, new claim for or significant modification to an
existing product, we must first receive either clearance under Section 510(k) of the Federal Food Drug and Cosmetic Act
("FDCA") marketing authorization under the de novo classification pathway, or approval of a PMA, from the FDA, unless
an exemption applies. In the 510(k) clearance process, before a device may be marketed, the FDA must determine that a
proposed device is “substantially equivalent” to a legally-marketed “predicate” device, which includes a device that has
been previously cleared through the 510(k) process, a device that was legally marketed prior to May 28, 1976 (pre-
amendments device), a device that was originally on the U.S. market pursuant to an approved PMA and later down-
classified, or a 510(k)-exempt device. To be “substantially equivalent,” the proposed device must have the same
intended use as the predicate device, and either have the same technological characteristics as the predicate device or
have different technological characteristics and not raise different questions of safety or effectiveness than the
predicate device. FDA then determines whether the device is as safe and effective as the predicate device by reviewing
the scientific methods used to evaluate differences in technological characteristics and performance data. Clinical data
are sometimes required to support substantial equivalence. In the process of obtaining PMA approval, the FDA must
determine that a proposed device is safe and effective for its intended use based, in part, on extensive data, including,
but not limited to, technical, pre-clinical, clinical trial, manufacturing and labeling data. The PMA process is typically
required for devices that are deemed to pose the greatest risk, such as life-sustaining, life-supporting or implantable
devices.
If no legally marketed predicate can be identified for a new device to enable use of the 510(k) pathway, the device is
automatically classified as Class III, which generally requires PMA approval. This results in some low to moderate risk
devices being classified unnecessarily as Class III. To address this, FDA regulations include a pathway to market for low
to moderate risk medical devices that are automatically placed into Class III due to the absence of a predicate device,
called the “Request for Evaluation of Automatic Class III Designation,” or the de novo classification procedure and
pathway. This procedure allows a manufacturer whose novel device is automatically classified into Class III to request
down-classification of its medical device into Class I or Class II on the basis that the device presents low or moderate
risk, rather than requiring the submission and approval of a PMA. The FDA can also reclassify a product from Class III to
Class I or II. If the data and information provided to the FDA in support of a de novo request demonstrate that general
controls or general and special controls are adequate to provide reasonable assurance of safety and effectiveness, and
the probable benefits of the device outweigh the probable risks, then the FDA will grant the de novo request authorizing
marketing of the product subject to the specified controls. After a device receives de novo classification, any
modification that could significantly affect its safety or efficacy, or that would constitute a major change or modification
in its intended use, will require a new 510(k) clearance or, depending on the modification, another de novo request or
even PMA approval.
Modifications to products that are approved through a PMA application generally require FDA approval. Similarly, certain
modifications made to products cleared through a 510(k) may require a new 510(k) clearance. Both the PMA approval
and the 510(k) clearance process can be expensive, lengthy and uncertain. The FDA’s 510(k) clearance process usually
takes from three to 12 months, but can last longer. The process of obtaining a PMA is generally much more costly and
uncertain than the 510(k) clearance process and generally takes from one to three years, or even longer, from the time
the application is submitted to the FDA. In addition, a PMA generally requires the performance of one or more clinical
trials. Despite the time, effort and cost, a device may not be approved or cleared by the FDA. Any delay or failure to
obtain necessary regulatory clearances or approvals could harm our business. Furthermore, even if we are granted
regulatory clearances or approvals, they may include significant limitations on the indicated uses for the device or other
restrictions or requirements, which may limit the market for the device.
33
The FDA, comparable foreign regulatory authorities or notified bodies can delay, limit or deny clearance, approval or
certification of a medical device for many reasons, including:
•our inability to demonstrate to the satisfaction of the FDA or the applicable regulatory authority or notified body
that our product candidates are safe or effective for their intended uses or are substantially equivalent to a
predicate device;
•the disagreement of the FDA or the applicable foreign regulatory authority with the design or implementation of
our clinical studies or the interpretation of data from pre-clinical studies or clinical studies;
•serious and unexpected adverse events experienced by participants in our clinical studies;
•the data from our pre-clinical studies and clinical studies may be insufficient to support clearance, approval or
certification where required;
•our inability to demonstrate that the clinical and other benefits of the device outweigh the risks;
•the manufacturing process or facilities we use may not meet applicable requirements; and
•the potential for approval policies or regulations of the FDA or applicable foreign regulatory authorities to change
significantly in a manner rendering our clinical data or regulatory filings insufficient for clearance or approval.
Subject to the transitional provisions and in order to sell our products in EU member states, our products must also
comply with the general safety and performance requirements of the EU Medical Devices Regulation, which repeals and
replaces the Medical Devices Directive. Compliance with these requirements is a prerequisite to be able to affix the
European Conformity ("CE") mark to our products, without which they cannot be sold or marketed in the European Union.
All medical devices placed on the market in the European Union must meet the general safety and performance
requirements laid down in Annex I to the EU Medical Devices Regulation including the requirement that a medical device
must be designed and manufactured in such a way that, during normal conditions of use, it is suitable for its intended
purpose. Medical devices must be safe and effective and must not compromise the clinical condition or safety of
patients, or the safety and health of users and – where applicable – other persons, provided that any risks which may be
associated with their use constitute acceptable risks when weighed against the benefits to the patient and are
compatible with a high level of protection of health and safety, taking into account the generally acknowledged state of
the art. Even if regulatory clearance, approval or certification is obtained, such products will remain subject to extensive
regulatory requirements. If we fail to comply with the regulatory requirements of the FDA and other applicable U.S. and
foreign regulatory authorities, or previously unknown problems with any approved commercial products, manufacturers
or manufacturing processes are discovered, we could be subject to administrative or judicially imposed sanctions. In
addition, the cost of compliance with new laws or regulations governing our technology or future products could
adversely affect our business, financial condition, results of operations and prospects. New laws or regulations may
impose restrictions or obligations on us that could force us to redesign our technology or other future products or
services, and may impose restrictions that are not possible or practicable to comply with, which could cause our
business to fail.
Our Commercial Products and any of our product candidates for which we obtain marketing approval in the future are
subject to post-marketing regulatory requirements and could be subject to post-marketing restrictions or withdrawal
from the market, and we may be subject to substantial penalties if we fail to comply with regulatory requirements or if
we experience unanticipated problems with our products following approval.
Once marketing approval has been granted, an approved product and its manufacturer and marketer are subject to
ongoing review and extensive regulation. Our Commercial Products and any of our product candidates for which we
obtain marketing clearance or approval in the future, as well as the manufacturing processes, post-approval studies and
measures, labeling, advertising and promotional activities for such products, among other things, will be subject to
continual requirements of and review by the FDA and other U.S. and foreign regulatory authorities. These requirements
include submissions of safety and other post-marketing information and reports, registration and listing requirements,
requirements relating to manufacturing, quality control, quality assurance and corresponding maintenance of records and
documents, and related compliance requirements such as price reporting, transparency reporting and requirements
regarding the distribution of samples to physicians and recordkeeping. Even if marketing authorization is granted, it may
be subject to limitations on the indicated uses for which the product may be marketed or to the conditions of approval,
including in the case of drug or biological products, the requirement to implement a REMS, which could include
requirements for a restricted distribution system.
The FDA and comparable foreign regulatory authorities may also impose requirements for costly post-marketing studies
or clinical trials and surveillance to monitor the safety or efficacy of a drug or biological product. There are similar
potential requirements for medical devices. In addition, manufacturers of approved products and those manufacturers’
facilities are required to comply with extensive requirements by the FDA and comparable foreign regulatory authorities,
including ensuring that quality control and manufacturing procedures conform to cGMP regulations, which include
requirements relating to quality control and quality assurance as well as the corresponding maintenance of records and
documentation and reporting requirements. We and our contract manufacturers could be subject to periodic
unannounced inspections by the FDA or foreign regulatory authorities to monitor and ensure compliance with cGMPs
(and similar foreign requirements) or other regulations.
34
If the FDA or another regulatory authority discovers previously unknown problems with a product, such as adverse
events of unanticipated severity or frequency, or problems with the facility where the product is manufactured, or
disagrees with the promotion, marketing or labeling of a product, such regulatory authorities may impose restrictions on
that product or us, including requiring withdrawal of the product from the market. If we fail to comply with applicable
regulatory requirements, a regulatory authority or enforcement authority may, among other things:
•refuse to approve pending applications or supplements to approved applications;
•require us to change the way a product is distributed, conduct additional clinical trials, change the labeling of a
product or require us to conduct additional post-marketing studies or surveillance;
•restrict our ability to conduct clinical trials, including full or partial clinical holds on ongoing or planned trials;
•require additional warnings on the product label, such as a “black box” warning, precaution or a contraindication;
•impose restrictions on the products, manufacturers or manufacturing process;
•require warning or untitled letters;
•seek injunctions or civil or criminal penalties;
•suspend or withdraw regulatory approvals;
•seize or detain products or implement import bans;
•impose voluntary or mandatory product recalls and publicity requirements;
•totally or partially suspend production; and
•impose restrictions on operations, including costly new manufacturing requirements.
Any government investigation of alleged violations of law could require us to expend significant time and resources in
response and could generate negative publicity. Any failure to comply with ongoing regulatory requirements may
adversely affect our ability to commercialize and generate revenue from our products. If regulatory sanctions are applied
or if regulatory approval is withdrawn, our business will be seriously harmed.
In connection with our Commercial Products and assuming we receive marketing approval for one or more of our product
candidates, we and our contract manufacturers will continue to expend time, money and effort in all areas of regulatory
compliance, including manufacturing, production, product surveillance and quality control. If we are not able to comply
with post-approval regulatory requirements, our ability to market any future products could be limited, which could
adversely affect our ability to sustain profitability. Further, the cost of compliance with post-approval regulations may
have a negative effect on our operating results and financial condition.
The FDA and other regulatory agencies actively enforce the laws and regulations prohibiting the promotion of
products inconsistent with approved labeling. If we are found or alleged to have improperly promoted a product, we
may become subject to significant liability.
The FDA and other U.S. or foreign agencies, including the U.S. Department of Justice ("DOJ") closely regulate and
monitor the post-approval marketing and promotion of drugs and biological products to ensure that they are
manufactured, marketed and distributed only for the approved indications and in accordance with the provisions of the
approved labeling. The FDA imposes stringent restrictions on manufacturers’ communications regarding off-label use,
and if we communicate about any of our product candidates for which we, or they, receive marketing approval in a way
that regulators assert goes beyond their approved indications, we, or they, may be subject to warnings or enforcement
action for off-label marketing. Alleged violations of the FDCA or other statutes, including the False Claims Act ("FCA")
relating to the promotion and advertising of prescription drugs may lead to investigations or allegations of violations of
federal and state health care fraud and abuse laws and state consumer protection laws.
In September 2021, the FDA published a Final Rule which describes the types of evidence that the agency will consider in
determining the intended use of a drug or biologic.
Notwithstanding the regulatory restrictions on off-label promotion, the FDA and other regulatory authorities allow
companies to engage in truthful, non-misleading, and non-promotional scientific communications concerning their
products in certain circumstances. For example, in January 2025, the FDA published final guidance outlining the agency’s
non-binding policies governing the distribution of scientific information on unapproved uses to healthcare providers. This
final guidance calls for such communications to be truthful, non-misleading, factual, and unbiased and include all
information necessary for healthcare providers to interpret the strengths and weaknesses and validity and utility of the
information about the unapproved use. In addition, under some relatively recent guidance from the FDA and the Pre-
Approval Information Exchange Act ("PIE Act") signed into law as part of the Consolidated Appropriations Act of 2023,
companies may also promote information that is consistent with the prescribing information and proactively speak to
formulary committee members of payors regarding data for an unapproved drug or unapproved uses of an approved
35
drug. We may engage in these discussions and communicate with healthcare providers, payors and other constituencies
in compliance with all applicable laws, regulatory guidance and industry best practices. We will need to carefully navigate
the FDA’s various regulations, guidance and policies, along with recently enacted legislation, to ensure compliance with
restrictions governing promotion of our Commercial Products.
In recent years, a significant number of pharmaceutical and biotechnology companies have been the target of inquiries
and investigations by various federal and state regulatory, investigative, prosecutorial and administrative entities in
connection with the promotion of products for unapproved uses and other sales practices, including the Department of
Justice and various U.S. Attorneys’ Offices, the Office of Inspector General of the Department of Health and Human
Services, the FDA, the Federal Trade Commission ("FTC") and various state Attorneys General offices. These
investigations have alleged violations of various federal and state laws and regulations, including claims asserting
antitrust violations, violations of the FDCA, the False Claims Act, the Prescription Drug Marketing Act and anti-kickback
laws and other alleged violations in connection with the promotion of products for unapproved uses, pricing and
Medicare and/or Medicaid reimbursement. Many of these investigations originate as “qui tam” actions under the False
Claims Act. Under the False Claims Act, any individual can bring a claim on behalf of the government alleging that a
person or entity has presented a false claim or caused a false claim to be submitted to the government for payment. The
person bringing a qui tam suit is entitled to a share of any recovery or settlement. Qui tam suits, also commonly referred
to as “whistleblower suits,” are often brought by current or former employees. In a qui tam suit, the government must
decide whether to intervene and prosecute the case. If it declines, the individual may pursue the case alone.
If the FDA or any other governmental agency initiates an enforcement action against us or if we are the subject of a qui
tam suit and it is determined that we violated prohibitions relating to the promotion of products for unapproved uses, we
could be subject to substantial civil or criminal fines or damage awards and other sanctions such as consent decrees and
corporate integrity agreements pursuant to which our activities would be subject to ongoing scrutiny and monitoring to
ensure compliance with applicable laws and regulations. Any such fines, awards or other sanctions would have an
adverse effect on our revenue, business, financial prospects and reputation.
Similar to the U.S., pharmaceutical companies in the European Union, the United Kingdom, and Australia are generally
prohibited from promoting drugs for any use that is not consistent with the product’s authorized uses. Doing so may
result in regulatory enforcement action. While off‑label prescribing by clinicians is permitted as part of medical practice,
the active promotion of off‑label uses by manufacturers is not.
In the European Union, promotion must be consistent with the approved marketing authorization. Promotion that
suggests or encourages use of a medicine for an unapproved indication, in an unapproved patient population, or using an
unapproved dose, route or regimen is treated as unlawful off‑label promotion. EU law distinguishes between the freedom
of healthcare professionals to prescribe off‑label based on clinical judgment and the strict limits on companies’
promotional activities, which may not extend beyond the authorized product information. Limited scientific exchange -
such as publication of data in peer‑reviewed journals or responses to unsolicited requests from healthcare professionals
- may be acceptable, but only where it is clearly non‑promotional, balanced, and not designed to increase prescribing.
In the United Kingdom, post‑Brexit rules continue to mirror this approach. Advertising of medicines is regulated by the
Human Medicines Regulations, enforced by the MHRA, and supplemented by the self‑regulatory Association of the
British Pharmaceutical Industry ("ABPI") Code. Both require that promotional materials align with the product’s current
labeling. Communications that directly or indirectly promote an unapproved indication or off‑label use breach these rules.
The MHRA and the Prescription Medicines Code of Practice Authority ("PMCPA"), which administers the ABPI Code,
routinely treat off‑label promotion as a serious violation. As in the European Union, manufacturers may provide scientific,
non‑promotional information—particularly in response to unsolicited, specific questions from healthcare professionals—
but must avoid any appearance of encouraging off‑label prescribing.
In Australia, the TGA, together with industry codes (such as the Medicines Australia Code of Conduct), prohibit
advertising of unapproved medicines and unapproved indications. Any communication that constitutes “advertising” and
promotes an off‑label use is not permitted, even if directed only to healthcare professionals. Promotional materials, detail
aids, and speaker programs must therefore be confined to the indications, populations and regimens approved in the
Australian Register of Therapeutic Goods ("ARTG") entry and associated product information. As in the European Union
and United Kingdom, companies may engage in limited scientific exchange, but this must be clearly separated from
promotional activities and must not function as de facto marketing for off‑label uses.
We may seek certain designations for our product candidates in the U.S., including breakthrough therapy, fast track
and priority review designations, and PRIME designation in the European Union, but we might not receive such
designations, and even if we do, such designations may not lead to a faster development or regulatory review or
approval process.
We may seek certain designations for one or more of our product candidates that could expedite review and approval by
the FDA. A breakthrough therapy-designated product candidate is defined as a product candidate that is intended, alone
or in combination with one or more other products, to treat a serious or life-threatening disease or condition, and
preliminary clinical evidence indicates that the product may demonstrate substantial improvement over existing therapies
on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical
development. For product candidates that have been designated as breakthrough therapies, interaction and
communication between the FDA and the sponsor of the trial can help to identify the most efficient path for clinical
development while minimizing the number of patients placed in ineffective control regimens. Breakthrough therapy
36
designation may not lead to a faster development or regulatory review or approval process, and does not increase the
likelihood that any product candidate that receives a breakthrough therapy designation will receive marketing approval.
The FDA may also issue fast track designation to a product candidate if it is intended, alone or in combination with one or
more other products, for the treatment of a serious or life-threatening disease or condition, and it demonstrates the
potential to address unmet medical needs for such a disease or condition. For fast track-designated product candidates,
sponsors may have greater interactions with the FDA and the FDA may initiate review of sections of a fast track product
candidate’s application before the application is complete. This rolling review may be available if the FDA determines,
after preliminary evaluation of clinical data submitted by the sponsor, that a fast track product may be effective.
We may also seek priority review for one or more of our product candidates. If the FDA determines that a product
candidate has the potential to provide a significant improvement in the treatment, diagnosis or prevention of a serious
disease or condition, and if approved would provide a significant improvement in the safety or effectiveness of the
treatment, prevention, or diagnosis of such disease or condition, the FDA may designate the product candidate for
priority review upon submission of a marketing application seeking approval of that product. A priority review designation
means that the goal for the FDA to review an application is six months, rather than the standard review period of ten
months.
These designations are within the discretion of the FDA. Accordingly, even if we believe that one of our product
candidates meets the criteria for these designations, the FDA may disagree and reject our request for designation.
Further, even if we receive a designation, the receipt of such designation for a product candidate may not result in a
faster development or regulatory review or approval process compared to product candidates considered for approval
under conventional FDA procedures, and the designation does not assure ultimate approval by the FDA. In addition, even
if one or more of our product candidates qualifies for these designations, the FDA may later decide that the product
candidates no longer meet the conditions for qualification and rescind the designation or decide that the time period for
FDA review or approval will not be shortened.
In the European Union, we may seek PRIME designation for some of our product candidates in the future. PRIME is a
voluntary program aimed at enhancing the EMA’s role to reinforce scientific and regulatory support in order to optimize
development and enable accelerated assessment of new medicines that are of major public health interest with the
potential to address unmet medical needs. The program focuses on medicines that target conditions for which there
exists no satisfactory method of treatment in the European Union or even if such a method exists, it may offer a major
therapeutic advantage over existing treatments. PRIME is limited to medicines under development and not authorized in
the European Union and the sponsor intends to apply for an initial MAA through the centralized procedure. To be
accepted for PRIME, a product candidate must meet the eligibility criteria with respect to its major public health interest
and therapeutic innovation based on information that is capable of substantiating the claims. The benefits of a PRIME
designation include the appointment of a CHMP rapporteur to provide continued support and help to build knowledge
ahead of a MAA, early dialogue and scientific advice at key development milestones, and the potential to qualify
products for accelerated review, meaning reduction in the review time for an opinion on approvability to be issued earlier
in the application process. PRIME enables a sponsor to request parallel EMA scientific advice and health technology
assessment advice to facilitate timely market access. Even if we or our collaborators receive PRIME designation for any
of our product candidates, the designation may not result in a materially faster development process, review or approval
compared to conventional EMA procedures. Further, obtaining PRIME designation does not assure or increase the
likelihood of the EMA’s grant of a marketing authorization.
We may submit an application for a Commissioner’s National Priority Voucher ("CNPV") from the FDA but there can be
no assurance that we will receive a voucher and, if a voucher is received, we may not be able to comply with the
requirements of the program. It is also possible that a competitor may receive a voucher which could harm our
competitive position in the marketplace.
In June 2025, the FDA announced the CNPV pilot program which was designed to accelerate the development and
review of certain drugs and biologics that are aligned with U.S. national health priorities, such as addressing a U.S. public
health crisis, developing more innovative cures for the American people, addressing a large unmet medical need,
onshoring drug development and manufacturing to advance the health interests of Americans and strengthen U.S. supply
chain resiliency, and increasing affordability. The FDA has stated that voucher recipients will receive a decision with
respect to an application on an accelerated basis, as well as enhanced communication with review staff throughout the
review process. The FDA expects the CNPV program to accelerate the application review timeline from 10-12 months to
1-2 months by convening a multidisciplinary team of physicians and scientists for a team-based review, interacting
frequently with the sponsor of the application to clarify questions, and completing review of the application concurrently.
The FDA retains full discretion to extend the review window if the data or application components submitted are
insufficient or incomplete, if the results of the pivotal trial(s) are ambiguous, or if the review is particularly complex. The
FDA has indicated that the CNPV program does not change the FDA’s rigorous safety and efficacy standards for review
and approval. In late 2025, FDA began issuing approvals for drugs with CNPVs within the 1-2 month review timeframe.
The CNPV program is new, limited in scope, and subject to evolving guidance, and available FDA resources. The FDA
retains broad discretion to modify the criteria, processes, or benefits of the program and may rescind participation or
alter timelines or the intended benefits at any time. Adding to the uncertainty, concerns have been raised regarding the
legality of the CNPV program.
We may seek approval of our product candidates from the FDA or comparable foreign regulatory authorities through
the use of accelerated development pathways. If we are not able to use such pathways, we may be required to
conduct additional clinical trials beyond those that are contemplated, which would increase the expense of obtaining,
37
and delay or prevent the receipt of, necessary marketing approvals. Moreover, even if we receive accelerated
approval from the FDA or comparable foreign regulatory authorities, if our confirmatory trials do not verify clinical
benefit, or if we do not comply with rigorous post-marketing requirements, the FDA or comparable foreign regulatory
authorities may seek to withdraw accelerated approval.
Under the Food, Drug, and Cosmetic Act ("FDCA"), and implementing regulations, the FDA may grant accelerated
approval to a product candidate designed to treat a serious or life-threatening condition that provides meaningful
therapeutic benefit over available therapies upon a determination that the product candidate has an effect on a
surrogate endpoint or intermediate clinical endpoint that is reasonably likely to predict clinical benefit. The FDA considers
a clinical benefit to be a positive therapeutic effect that is clinically meaningful in the context of a given disease, such as
irreversible morbidity or mortality. For the purposes of accelerated approval, a surrogate endpoint is a marker, such as a
laboratory measurement, radiographic image, physical sign, or other measure that is thought to predict clinical benefit,
but is not itself a measure of clinical benefit. An intermediate clinical endpoint is a clinical endpoint that can be measured
earlier than an effect on irreversible morbidity or mortality that is reasonably likely to predict an effect on irreversible
morbidity or mortality or other clinical benefit measurement of a therapeutic effect that is considered reasonably likely to
predict the clinical benefit. Prior to seeking such accelerated approval, we will continue to seek feedback from the FDA
or comparable foreign regulatory agencies and otherwise evaluate our, or their, ability to seek and receive such
accelerated approval.
There can be no assurance that the FDA or foreign regulatory agencies will agree with our surrogate endpoints or
intermediate clinical endpoints in any of our clinical trials, or that we will decide to pursue or submit any additional NDAs
or BLAs seeking accelerated approval. Similarly, there can be no assurance that, after feedback from the FDA or
comparable foreign regulatory agencies, we will continue to pursue or apply for accelerated approval. Furthermore, for
any submission of an application for accelerated approval, there can be no assurance that such submission will be
accepted for filing or that any expedited development, review or approval will be granted on a timely basis, or at all.
Finally, there can be no assurance that we will satisfy all FDA requirements, including new provisions that govern
accelerated approval. For example, with passage of the FDORA in December 2022, Congress modified certain provisions
governing accelerated approval of drug and biologic products. Specifically, the new legislation (i) authorized FDA to
require a sponsor to have its confirmatory clinical trial underway before accelerated approval is awarded; (ii) requires a
sponsor of a product granted accelerated approval to submit progress reports on its post-approval studies to FDA every
six months until the study is completed; and (iii) authorizes FDA to use expedited procedures to withdraw accelerated
approval of an NDA or a BLA if certain conditions are met, including where a required confirmatory study fails to verify
and describe the predicted clinical benefit or where evidence demonstrates the product is not shown to be safe or
effective under the conditions of use. The FDA may also use such procedures to withdraw an accelerated approval if a
sponsor fails to conduct any required post-approval study of the product with due diligence, including with respect to
“conditions specified by the Secretary.” The new procedures include the provision of due notice and an explanation for a
proposed withdrawal, and opportunities for a meeting with the Commissioner or the Commissioner’s designee and a
written appeal, among other things. We will need to fully comply with these and other requirements in connection with
the development and approval of any product candidate that qualifies for accelerated approval.
In March 2023, the FDA issued draft guidance that outlines its current thinking and approach to accelerated approval.
The FDA indicated that the accelerated approval pathway is commonly used for approval of oncology drugs due to the
serious and life-threatening nature of cancer. Although single-arm trials have been commonly used to support
accelerated approval, a randomized controlled trial is the preferred approach as it provides a more robust efficacy and
safety assessment and allows for direct comparisons to an available therapy. To that end, the FDA outlined
considerations for designing, conducting, and analyzing data for trials intended to support accelerated approvals of
oncology therapeutics. Subsequently, in December 2024 and January 2025, the FDA issued additional draft guidances
relating to accelerated approval. These guidances describe FDA’s latest thinking on what it means to conduct a
confirmatory trial with due diligence and how the agency plans to interpret whether such a study needs to be underway
at the time of approval. While these guidances are currently only in draft form and will ultimately not be legally binding
even when finalized, sponsors typically observe the FDA’s guidance closely to ensure that their investigational products
qualify for accelerated approval.
In the European Union, a “conditional” marketing authorization may be granted in cases where all the required safety and
efficacy data are not yet available. A conditional marketing authorization is subject to conditions to be fulfilled for
generating missing data or ensuring increased safety measures. A conditional marketing authorization is valid for one
year and has to be renewed annually until fulfillment of all relevant conditions. Once the applicable pending studies are
provided, a conditional marketing authorization can become a “standard” marketing authorization. However, if the
conditions are not fulfilled within the timeframe set by the EMA, the marketing authorization will cease to be renewed.
Accordingly, a failure to obtain and maintain accelerated approval or any other form of expedited development, review or
approval for our product candidates, or withdrawal of a product candidate, would result in a longer time period until
commercialization of such product candidate, could increase the cost of development of such product candidate and
could harm our competitive position in the marketplace.
We may not be able to obtain orphan drug designation or exclusivity for any product candidates we may develop, and
even if we do, that exclusivity may not prevent the FDA or foreign regulatory authorities from approving other
competing products.
38
Under the Orphan Drug Act, the FDA may designate a product as an orphan drug if it is a drug or biologic intended to
treat a rare disease or condition, meaning that the product is intended for a condition or disease with a patient population
of fewer than 200,000 individuals annually in the U.S., or more than 200,000 individuals in the U.S. for which there is no
reasonable expectation that the cost of developing and making available in the U.S. the drug or biologic will be recovered
from sales in the U.S. for that drug or biologic. For example, we have received orphan drug designations from the FDA for
TLX101-Tx for the treatment of glioma, for TLX101-Px for the imaging of glioma and for TLX66-Tx as a conditioning
treatment prior to hematopoietic stem cell transplant. TLX090-Tx has also been granted orphan drug designation by the
FDA for the treatment of osteosarcoma, and TLX102-Tx has been granted orphan drug designation for the treatment of
multiple myeloma and malignant glioma. In addition, in the European Union, a medicinal product may be designated as
orphan if its sponsor can establish that (i) the product is intended for the diagnosis, prevention or treatment of a life-
threatening or chronically debilitating condition; (ii) either (a) such condition affects no more than 5 in 10,000 persons in
the European Union when the application is made, or (b) the product, without the benefits derived from orphan status,
would not generate sufficient return in the European Union to justify investment; and (iii) there exists no satisfactory
method of diagnosis, prevention or treatment of such condition authorized for marketing in the European Union, or if
such a method exists, the medicinal product will be of significant benefit to those affected by the condition. For example,
TLX101-Tx and TLX66-Tx have been granted orphan drug designation in Europe. Orphan drug designation may not lead
to a faster development or regulatory review or approval process and does not increase the likelihood that any product
candidate that receives an orphan drug designation will receive marketing approval.
Generally, if a product candidate with an orphan drug designation subsequently receives the first marketing approval for
the disease or condition for which it has such designation, the product is entitled to a period of marketing exclusivity,
which precludes the FDA or foreign regulatory authorities, as applicable, from approving another marketing application
for the same product for the same disease or condition for a prescribed time period. The applicable period is seven years
in the U.S. and ten years in the European Union. The exclusivity period in the European Union can be reduced to six years
if, at the end of the fifth year, a product no longer meets the criteria for Orphan Designation, in particular if the product is
sufficiently profitable so that market exclusivity is no longer justified. Even if we obtain the designation and if, upon
approval, we obtain orphan drug exclusivity for a product, that exclusivity may not effectively protect the product from
competition because different products can be approved for the same disease or condition. In addition, even after an
orphan drug or biologic is approved, the FDA and comparable foreign regulatory authorities, such as the European
Commission, can subsequently approve the same product for the same condition if the FDA or such other authorities
conclude that the later product is clinically superior in that it is shown to be safer, more effective or makes a major
contribution to patient care. Orphan drug exclusivity may also be lost if the FDA or comparable foreign regulatory
authorities determines that the request for designation was materially defective or if the manufacturer is unable to assure
sufficient quantity of the product to meet the needs of the patients with the rare disease or condition.
The FDA and Congress may further reevaluate the Orphan Drug Act and its regulations and policies. This may be
particularly true in light of a decision from the Court of Appeals for the 11th Circuit in September 2021 finding that, for the
purpose of determining the scope of exclusivity, the term “same disease or condition” means the designated “rare
disease or condition” and could not be interpreted by the FDA to mean the “indication or use.” Thus, the court concluded,
orphan drug exclusivity applies to the entire designated disease or condition rather than the “indication or use.” Although
there have been legislative proposals to overrule this decision, they have not been enacted into law. On January 23,
2023, the FDA announced that, in matters beyond the scope of that court order, the FDA will continue to apply its
existing regulations tying orphan-drug exclusivity to the uses or indications for which the orphan drug was approved. We
do not know if, when, or how the FDA may change the orphan drug regulations and policies in the future or whether
Congress will take legislative action, and it is uncertain how any changes might affect our business. Depending on what
changes the FDA or Congress may make to orphan drug regulations and policies, our business could be adversely
impacted.
If the FDA does not conclude that certain of our product candidates satisfy the requirements for the Section
505(b)(2) regulatory approval pathway, or if the requirements for such product candidates under Section 505(b)(2)
are not as we expect, the approval pathway for those product candidates will likely take significantly longer, cost
significantly more and entail significantly greater complications and risks than anticipated, and in either case may not
be successful.
We are developing certain product candidates for which we may seek FDA approval through the Section 505(b)(2)
regulatory pathway. The Drug Price Competition and Patent Term Restoration Act of 1984 ("the Hatch-Waxman Act")
added Section 505(b)(2) to the FDCA. Section 505(b)(2) permits the filing of an NDA where at least some of the
information required for approval comes from studies that were not conducted by or for the applicant and for which the
applicant has not obtained a right of reference. Section 505(b)(2), if applicable to us under the FDCA, would allow an
NDA we submit to the FDA to rely in part on data in the public domain or the FDA’s prior conclusions regarding the safety
and effectiveness of approved compounds, which could expedite the development program for our product candidates
by potentially decreasing the amount of clinical data that we would need to generate in order to obtain FDA approval.
If the FDA does not allow us to pursue the Section 505(b)(2) regulatory pathway as anticipated, we may need to conduct
additional clinical trials, provide additional data and information, and meet additional standards for regulatory approval.
If this were to occur, the time and financial resources required to obtain FDA approval for these product candidates, and
complications and risks associated with these product candidates, would likely substantially increase. We could need to
obtain additional funding, which could result in significant dilution to the ownership interests of our then existing
shareholders to the extent we issue equity securities or convertible debt. We cannot guarantee that we would be able to
39
obtain such additional financing on terms acceptable to us, if at all. Moreover, inability to pursue the Section 505(b)(2)
regulatory pathway would likely result in new competitive products reaching the market more quickly than our product
candidates, which would likely materially adversely impact our competitive position and prospects. Even if we are
allowed to pursue the Section 505(b)(2) regulatory pathway, we cannot guarantee that our product candidates will
receive the requisite approvals for commercialization.
In addition, notwithstanding the approval of a number of products by the FDA under Section 505(b)(2) over the last few
years, certain brand-name pharmaceutical companies and others have objected to the FDA’s interpretation of Section
505(b)(2). If the FDA’s interpretation of Section 505(b)(2) is successfully challenged, the FDA may change its 505(b)(2)
policies and practices, which could delay or even prevent the FDA from approving any NDA that we submit under Section
505(b)(2). In addition, the pharmaceutical industry is highly competitive, and Section 505(b)(2) NDAs are subject to
special requirements designed to protect the patent rights of sponsors of previously approved drugs that are referenced
in a Section 505(b)(2) NDA. These requirements may give rise to patent litigation and mandatory delays in approval of
our NDAs for up to 30 months or longer depending on the outcome of any litigation. It is not uncommon for a
manufacturer of an approved product to file a citizen petition with the FDA seeking to delay approval of, or impose
additional approval requirements for, pending competing products. If successful, such petitions can significantly delay, or
even prevent, the approval of the new product. However, even if the FDA ultimately denies such a petition, the FDA may
substantially delay approval while it considers and responds to the petition. In addition, even if we are able to utilize the
Section 505(b)(2) regulatory pathway, there is no guarantee this would ultimately lead to accelerated product
development or earlier approval.
Moreover, even if our product candidates are approved under Section 505(b)(2), the approval may be subject to
limitations on the indicated uses for which the products may be marketed or to other conditions of approval, or may
contain requirements for costly post-marketing testing and surveillance to monitor the safety or efficacy of the products.
Disruptions at the FDA, the SEC and other government agencies, including from inadequate funding, government
shut downs, reductions in force, or other disruptions to these agencies’ operations, could hinder their ability to hire
and retain key leadership and other personnel, prevent new products and services from being developed or
commercialized in a timely manner, if at all, or otherwise prevent those agencies from performing normal business
functions on which the operation of our business may rely, which could negatively impact our business. In addition,
changes in FDA policies or regulations, resulting from the foregoing disruptions or otherwise, could adversely impact
the development of our product candidates and, ultimately, our ability to receive approval for and commercialize
them.
The ability of the FDA and comparable foreign regulatory authorities (or notified bodies) to review and approve or certify
new products can be affected by a variety of factors, including government budget and funding levels, reductions in
force, government shutdowns, ability to hire and retain key personnel and accept the payment of user fees, and
statutory, regulatory and policy changes. Average review times at the agency have fluctuated in recent years as a result.
Disruptions at the FDA, other agencies, and authorities (or notified bodies) may also slow the time necessary to meet
with and receive agency feedback, review and/or approve our submissions, conduct inspections, issue regulatory
guidance, or take other actions that facilitate the development, approval and marketing of regulated products, which
would adversely affect our business. Moreover, changes in FDA’s policies or regulations, whether as a result of personnel
and budgetary constraints described above, changes in leadership or otherwise, could adversely impact the
development of our product candidates and, ultimately, our ability to receive marketing authorization. Decisions about
the development of product candidates are often based on interactions with FDA and regulatory guidance provided by
the agency following such interactions. If FDA does not agree with our decisions resulting from such interactions and
guidance or there are subsequent policy changes, review and approval of our product candidates may be delayed or not
occur at all. In addition, government funding of the SEC and other government agencies on which our operations may
rely, including those that fund research and development activities, is subject to the political process, which is inherently
fluid and unpredictable.
Disruptions at the FDA, other agencies, and authorities (or notified bodies) may also slow the time necessary for new
product candidates to be reviewed and/or approved (or certified) by necessary government agencies, foreign regulatory
authorities (or notified bodies), which would adversely affect our business. For example, over the last several years the
U.S. government has shut down several times, including most recently in October and November 2025, and certain
regulatory agencies, such as the FDA and the SEC, have had to furlough critical employees and stop critical activities.
In addition, disruptions may result from events similar to the COVID-19 pandemic. During the COVID-19 pandemic, a
number of companies announced receipt of complete response letters due to the FDA’s inability to complete required
inspections for their applications. In the event of a similar public health emergency in the future, the FDA may not be able
to continue its current pace and review timelines could be extended. Regulatory authorities outside the U.S. facing similar
circumstances may adopt similar restrictions or other policy measures in response to a similar public health emergency
and may also experience delays in their regulatory activities.
If another prolonged government shutdown occurs, it could significantly impact the ability of the FDA to timely review
and process our regulatory submissions, which could have a material adverse effect on our business. Further, future
government shutdowns could impact our ability to access the public markets and obtain necessary capital in order to
properly capitalize and continue our operations.
40
Recently enacted and future legislation may increase the difficulty and cost for us to commercialize our or our
collaborators' product candidates, if approved, and affect the prices we, or they, may obtain.
In the U.S. and some foreign jurisdictions, there have been a number of legislative and regulatory changes and proposed
changes regarding the healthcare system that could, among other things, restrict or regulate post-approval activities and
affect our ability to profitably sell or commercialize our Commercial Products or any product candidate for which we, or
our collaborators, obtain marketing approval. We expect that current laws, as well as other healthcare reform measures
that may be adopted in the future, may result in more rigorous coverage criteria and in additional downward pressure on
the price that we, or any collaborators, may receive for any approved products. If reimbursement of our products is
unavailable or limited in scope, our business could be materially harmed.
Because our Commercial Products are generally reimbursed under Medicare Part B which covers drugs administered by
healthcare providers in outpatient settings, changes to CMS coverage, packaging or bundling policies and site‑of‑service
payment could directly affect utilization and net pricing. We expect that current laws, as well as other healthcare reform
measures that may be adopted in the future, may result in more rigorous coverage criteria and an additional downward
pressure on the price that we, or any collaborators, may receive for any approved products. If reimbursement of our
products is unavailable or limited in scope, our business could be materially harmed.
In March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education
Affordability Reconciliation Act (collectively "the ACA") was enacted. The ACA established an annual, nondeductible fee
on any entity that manufactures or imports specified branded prescription drugs and biologic agents; extended
manufacturers’ Medicaid rebate liability to covered drugs dispensed to individuals who are enrolled in Medicaid managed
care organizations; expanded eligibility criteria for Medicaid programs; expanded the entities eligible for discounts under
the 340B drug pricing program; increased the statutory minimum rebates a manufacturer must pay under the Medicaid
Drug Rebate Program; established a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in,
and conduct comparative clinical effectiveness research, along with funding for such research; and established a Center
for Medicare & Medicaid Innovation at CMS, an agency within the U.S. Department of Health and Human Services
("HHS") to test innovative payment and service delivery models to lower Medicare and Medicaid spending. Since its
enactment, there have been executive, judicial, and Congressional challenges to certain aspects of the ACA.
In addition, other legislative changes have been proposed and adopted since the ACA was enacted. In August 2011, the
Budget Control Act of 2011, among other things, created measures for spending reductions by Congress. A Joint Select
Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least US$1.2 trillion for the
years 2013 through 2021, was unable to reach required goals, thereby triggering the legislation’s automatic reduction to
several government programs. These changes included aggregate reductions to Medicare payments to providers, which
went into effect in April 2013 and will remain in effect through 2032. The American Taxpayer Relief Act of 2012, among
other things, reduced Medicare payments to several providers and increased the statute of limitations period for the
government to recover overpayments to providers from three to five years. Further, with the passage of the Inflation
Reduction Act ("IRA") in August 2022, Congress authorized Medicare drug price negotiation for certain high‑expenditure,
single‑source drugs and biologics (beginning with 10 Part D products in 2026 and increasing over time), imposed
Medicare Part B and Part D inflation rebates, and redesigned Part D to cap beneficiary out‑of‑pocket spending at
US$2,000 beginning in 2025.
In May 2025, President Trump issued an executive order implementing the concept of most-favored nation pricing.
Under this order, the Department of Health and Human Services, in coordination with other federal agencies, is directed
to take actions to ensure that the price of prescription drugs paid by federal health insurers, including Medicare and
Medicaid, is in line with the prices paid in comparably developed nations. It is unclear how this policy will impact our
business.
As an alternative to the Affordable Care Act, President Trump recently announced the Great Healthcare Plan. As
presented, the plan is intended to lower drug prices by increasing competition and benchmarking U.S. drug prices to
other countries, reduce insurance premiums by redirecting subsidies from insurers to individuals, increase accountability
and transparency from insurers, and promote consumer choice by giving individuals more direct control over how
healthcare dollars are spent. Legislative and regulatory action will be required to fully implement the plan. It is unclear
how these proposed changes will impact our business and the pharmaceutical industry in general.
These and other executive and legislative efforts may result in additional reductions in Medicare and other healthcare
funding and otherwise affect the prices we may obtain for any of our products or product candidates for which we may
obtain regulatory approval or the frequency with which any such product is prescribed or used. For example, on March
11, 2021, the American Rescue Plan Act of 2021 was signed into law, which eliminates the statutory cap on the Medicaid
drug rebate, beginning January 1, 2024. The rebate was previously capped at 100% of a drug’s average manufacturer
price.
In the European Union, on December 13, 2021, Regulation No 2021/2282 on Health Technology Assessment ("HTA")
amending Directive 2011/24/EU, was adopted. While the Regulation entered into force in January 2022, it began to apply
from January 2025 onwards and will have a phased implementation depending on the concerned products. The
Regulation intends to boost cooperation among EU member states in assessing health technologies, including new
medicinal products as well as certain high-risk medical devices, and provide the basis for cooperation at the EU level for
joint clinical assessments in these areas. It will permit EU member states to use common HTA tools, methodologies, and
procedures across the European Union, working together in four main areas, including joint clinical assessment of the
41
innovative health technologies with the highest potential impact for patients, joint scientific consultations whereby
developers can seek advice from HTA authorities, identification of emerging health technologies to identify promising
technologies early, and continuing voluntary cooperation in other areas. Individual EU member states will continue to be
responsible for assessing non-clinical (e.g., economic, social, ethical) aspects of health technology, and making decisions
on pricing and reimbursement.
We expect that these healthcare reforms, as well as other healthcare reform measures that may be adopted in the future,
may result in additional reductions in Medicare and other healthcare funding, more rigorous coverage criteria and new
payment methodologies that govern our Commercial Products or any product candidate, if approved, and/or the level of
reimbursement physicians receive for administering our Commercial Products or any other product candidate, if
approved, we might bring to market. Reductions in reimbursement levels may negatively impact the prices we receive or
the frequency with which our products are prescribed or administered. Any reduction in reimbursement from Medicare or
other government programs may result in a similar reduction in payments from private payors. Accordingly, such reforms,
if enacted, could have an adverse effect on anticipated revenue from our Commercial Products or from product
candidates for which we may obtain marketing approval and may affect our overall financial condition and ability to
develop or commercialize product candidates.
The insurance coverage and reimbursement status of newly approved products is uncertain. Our Commercial
Products and product candidates, if approved, may become subject to unfavorable pricing regulations, third-party
coverage and reimbursement practices, or healthcare reform initiatives, which would harm our business. Failure to
obtain or maintain coverage and adequate reimbursement for our Commercial Products or any product candidates
for which we obtain approval could limit our ability to market those products and decrease our ability to generate
revenue.
The regulations that govern marketing approvals, pricing, coverage, and reimbursement for new drugs and other medical
products vary widely from country to country. In the U.S., healthcare reform legislation may significantly change the
approval requirements in ways that could involve additional costs and cause delays in obtaining approvals. Some
countries require approval of the sale price of a product before it can be marketed. In many countries, the pricing review
period begins after marketing or product licensing approval is granted. In some foreign markets, pricing remains subject
to continuing governmental control even after initial approval is granted. As a result, we might obtain marketing approval
for a product in a particular country, but then be subject to price regulations that delay our commercial launch of the
product, possibly for lengthy time periods, and negatively impact the revenue we are able to generate from the sale of
the product in that country. Adverse pricing limitations may hinder our ability to recoup our investment in our Commercial
Products or product candidates, even if any product candidates we may develop obtain marketing approval.
Our ability to successfully commercialize our Commercial Products and product candidates, if approved, also will depend
in part on the extent to which coverage and adequate reimbursement for these products and related treatments will be
available from government health administration authorities, private health insurers, and other organizations. Government
authorities and third-party payors, such as private health insurers and health maintenance organizations, decide which
medications they will pay for and establish reimbursement levels. The availability of coverage and extent of
reimbursement by governmental and private payors is essential for most patients to be able to afford treatments. Sales
of our Commercial Products or product candidates, if approved, that we may identify will depend substantially, both
domestically and abroad, on the extent to which the costs of our Commercial Products and product candidates, if
approved, will be paid by health maintenance, managed care, pharmacy benefit and similar healthcare management
organizations, or reimbursed by government health administration authorities, private health coverage insurers and other
third-party payors. If coverage and adequate reimbursement is not available, or is available only to limited levels, we may
not be able to successfully commercialize our Commercial Products or product candidates, if approved. Even if coverage
is provided, the approved reimbursement amount may not be high enough to allow us to establish or maintain pricing
sufficient to realize a sufficient return on our investment. A primary trend in the U.S. healthcare industry and elsewhere is
cost containment. Government authorities and third-party payors have attempted to control costs by limiting coverage
and the amount of reimbursement for particular medications. In many countries, the prices of medical products are
subject to varying price control mechanisms as part of national health systems. In general, the prices of medicines under
such systems are substantially lower than in the U.S. Other countries allow companies to fix their own prices for
medicines, but monitor and control company profits. Additional foreign price controls or other changes in pricing
regulation could restrict the amount that we are able to charge for our products and product candidates. Accordingly, in
markets outside the U.S., the reimbursement for products may be reduced compared with the U.S. and may be
insufficient to generate commercially reasonable revenues and profits.
There is also significant uncertainty related to the insurance coverage and reimbursement of newly approved products
and coverage may be more limited than the purposes for which the medicine is approved by the FDA or comparable
foreign regulatory authorities. In the U.S., the principal decisions about reimbursement for new medicines are typically
made by CMS. CMS decides whether and to what extent a new medicine will be covered and reimbursed under Medicare
and private payors tend to follow CMS to a substantial degree. No uniform policy of coverage and reimbursement for
products exists among third-party payors and coverage and reimbursement levels for products can differ significantly
from payer to payer. As a result, the coverage determination process is often a time consuming and costly process that
may require us to provide scientific and clinical support for the use of our products to each payer separately, with no
assurance that coverage and adequate reimbursement will be applied consistently or obtained in the first instance. It is
difficult to predict what CMS will decide with respect to reimbursement for fundamentally novel products such as ours,
as there is no body of established practices and precedents for these new products.
42
Reimbursement agencies in Europe may be more conservative than CMS. For example, a number of cancer drugs have
been approved for reimbursement in the U.S. and have not been approved for reimbursement in certain European
countries. Moreover, eligibility for reimbursement does not imply that any drug will be paid for in all cases or at a rate that
covers our costs, including research, development, manufacture, sale, and distribution. Interim reimbursement levels for
new drugs, if applicable, may also not be sufficient to cover our costs and may not be made permanent.
Reimbursement rates may vary according to the use of the drug and the clinical setting in which it is used, may be based
on reimbursement levels already set for lower cost drugs and may be incorporated into existing payments for other
services. Our inability to promptly obtain coverage and profitable payment rates from both government-funded and
private payors for our Commercial Products or any approved products we may develop could have a material adverse
effect on our operating results, our ability to raise capital needed to commercialize product candidates, if approved, and
our overall financial condition. Further, the number of uninsured individuals in the U.S. may increase, which may
adversely affect our ability to commercialize our Commercial Products or any product candidates for which we receive
marketing authorization, In the U.S., we have various programs to help patients afford our Commercial Products,
including patient assistance programs and co-pay coupon programs for eligible patients.
Net prices for drugs may be reduced by mandatory discounts or rebates required by government healthcare programs or
private payors and by any future relaxation of laws that presently restrict imports of drugs from countries where they
may be sold at lower prices than in the U.S. Our inability to promptly obtain coverage and profitable reimbursement rates
from third-party payors for our Commercial Products any product candidates that we develop and for which we obtain
marketing authorization could have a material adverse effect on our operating results, our ability to raise capital needed
to commercialize products and our overall financial condition.
Increasingly, third-party payors are requiring that pharmaceutical companies provide them with predetermined discounts
from list prices and are challenging the prices charged for medical products. We cannot be sure that reimbursement will
be available for any product candidate for which we receive marketing authorization and that we commercialize and, if
reimbursement is available, the level of reimbursement. Reimbursement may impact the demand for, or the price of, any
product or product candidate for which we obtain marketing approval. In order to obtain reimbursement, physicians may
need to show that patients have superior treatment outcomes with our products compared to standard-of-care drugs,
including lower-priced generic versions of standard-of-care drugs. We expect to experience pricing pressures in
connection with the sale of our Commercial Products and our product candidates, if approved, due to the trend toward
managed healthcare, the increasing influence of health maintenance organizations and additional legislative changes.
The downward pressure on healthcare costs in general, particularly prescription drugs and surgical procedures and other
treatments, has become very intense. As a result, increasingly high barriers are being erected to the entry of new
products. Additionally, we may develop companion diagnostic tests for use with our therapeutic product candidates. We
may be required to obtain coverage and reimbursement for these tests separate and apart from the coverage and
reimbursement we seek for our product candidates, once approved. Even if we obtain regulatory approval or clearance
for such companion diagnostics, there is significant uncertainty regarding our ability to obtain coverage and adequate
reimbursement for the same reasons applicable to our product candidates. Medicare reimbursement methodologies,
whether under Part A, Part B, or clinical laboratory fee schedule may be amended from time to time, and we cannot
predict what effect any change to these methodologies would have on any product candidate or companion diagnostic
for which we receive approval.
The prices of prescription pharmaceuticals in the U.S. and foreign jurisdictions are subject to considerable legislative
and executive actions and could impact the prices we obtain for our products, if and when approved.
The prices of prescription pharmaceuticals have also been the subject of considerable discussion in the U.S.. There have
been several recent U.S. congressional inquiries, as well as proposed and enacted state and federal legislation designed
to, among other things, bring more transparency to pharmaceutical pricing, review the relationship between pricing and
manufacturer patient programs, and reduce the costs of pharmaceuticals under Medicare and Medicaid.
In October 2020, HHS and the FDA published a final rule allowing states and other entities to develop a Section 804
Importation Program ("SIP") to import certain prescription drugs from Canada into the U.S. Section 804 of the FDCA
allows importation of prescription drugs from Canada to significantly reduce the cost of these drugs to the American
consumer without imposing additional risk to public health and safety. Importation program proposals must be submitted
to FDA for review and authorization. Several states have passed laws allowing for the importation of drugs from Canada.
Several states have passed legislation establishing workgroups to examine the impact of a state importation program. In
May 2025, FDA announced that it will offer individual states the opportunity to submit draft proposals for pre-review and
obtain initial feedback from FDA prior to formally submitting their program proposal. States will have an opportunity to
meet with FDA with the goal of reducing the burden on the states and to help them develop robust proposals. FDA is also
working to assist states with options to streamline the required cost savings analysis, and to provide input regarding the
information the states may rely on to estimate cost savings for American consumers.
Further, on November 20, 2020, HHS finalized a regulation removing safe harbor protection for price reductions from
pharmaceutical manufacturers to plan sponsors under Part D, either directly or through pharmacy benefit managers,
unless the price reduction is required by law. The final rule would also eliminate the current safe harbor for Medicare
drug rebates and create new safe harbors for beneficiary point-of-sale discounts and pharmacy benefit manager service
fees. It was originally set to go into effect on January 1, 2022, but with passage of the IRA, has been delayed by
Congress to January 1, 2032.
43
On August 16, 2022, the IRA was enacted. The new legislation has implications for Medicare Part D, which is a program
available to individuals who are entitled to Medicare Part A or enrolled in Medicare Part B to give them the option of
paying a monthly premium for outpatient prescription drug coverage. Among other things, the IRA requires
manufacturers of certain drugs to engage in price negotiations with Medicare (beginning in 2026), with prices that can be
negotiated subject to a cap; imposes rebates under Medicare Part B and Medicare Part D to penalize price increases that
outpace inflation (which began in 2023); and replaces the Part D coverage gap discount program with a new discounting
program (which began in 2025). The IRA permits the Secretary of the HHS to implement many of these provisions
through guidance, as opposed to regulation, for the initial years.
Specifically, with respect to price negotiations, Congress authorized Medicare to negotiate lower prices for certain costly
single-source drug and biologic products that do not have competing generics or biosimilars and are reimbursed under
Medicare Part B and Part D. CMS may negotiate prices for ten high-cost drugs paid for by Medicare Part D starting in
2026, followed by 15 Part D drugs in 2027, 15 Part B or Part D drugs in 2028, and 20 Part B or Part D drugs in 2029 and
beyond. This provision applies to drug products that have been approved for at least nine years and biologics that have
been licensed for 13 years, but it does not apply to drugs and biologics that have been approved for a single rare disease
or condition. Further, the legislation subjects drug manufacturers to civil monetary penalties and a potential excise tax for
failing to comply with the legislation. The legislation also requires manufacturers to pay rebates for drugs in Medicare
Part D whose price increases exceed inflation. The law also capped Medicare out-of-pocket drug costs at approximately
US$4,000 a year in 2024 and, thereafter beginning in 2025, at US$2,000 a year (indexed for inflation in subsequent
years).
The first cycle of negotiations for the Medicare Drug Price Negotiation Program commenced in the summer of 2023. On
August 15, 2024, the HHS published the negotiated "maximum fair prices" for ten selected Part D drugs that treat a range
of conditions, including diabetes, chronic kidney disease, and rheumatoid arthritis. The prices of these ten drugs became
effective January 1, 2026. On January 17, 2025, CMS announced its selection of 15 additional drugs covered by Part D
for the second cycle of negotiations. The negotiated prices for this second group of drugs will be effective on January 1,
2027. While there had been some questions about the Trump Administration’s position on this program, on September
30, 2025, CMS issued final guidance for the third negotiation cycle. On January 27, 2026, CMS announced its selection
of 15 additional drugs (covered under either Part B or Part D) for the third cycle of negotiations. The negotiated prices for
this third group will be effective on January 1, 2028. CMS also selected one previously negotiated drug for the program’s
first renegotiations.
On June 6, 2023, Merck & Co. filed a lawsuit against the HHS and CMS asserting that, among other things, the IRA’s Drug
Price Negotiation Program for Medicare constitutes an uncompensated taking in violation of the Fifth Amendment of the
Constitution. Subsequently, a number of other parties, also filed lawsuits in various courts with similar constitutional
claims against the HHS and CMS. As of October 2025, Merck’s action is pending motions for summary judgment, and the
other lawsuits have been largely unsuccessful for plaintiffs. Multiple federal district courts have granted summary
judgment in favor of the HHS, and several of these decisions have been affirmed by federal appellate courts. For
example, in 2025, the U.S. Court of Appeals for the Third Circuit rejected appeals from several manufacturers, including
AstraZeneca and Novartis, upholding the program’s constitutionality. The Second and Sixth Circuits have also affirmed
dismissals of similar challenges. In September 2025, AstraZeneca filed a petition for writ of certiorari, asking the U.S.
Supreme Court to review the Third Circuit’s decision. Accordingly, while the IRA is actively being implemented, the
pending appeal to the Supreme Court creates uncertainty regarding the program’s ultimate legal status. We cannot
predict with certainty what impact any federal or state health reforms will have on us, but such changes could impose
new or more stringent regulatory requirements on our activities or result in reduced reimbursement for our products, any
of which could adversely affect our business, results of operations and financial condition.
In May 2025, President Trump issued an executive order implementing the concept of most-favored nation pricing.
Under this order, the Department of Health and Human Services, in coordination with other federal agencies, is directed
to take actions to ensure that the price of prescription drugs paid by federal health insurers, including Medicare and
Medicaid, is in line with the prices paid in comparably developed nations. It is unclear how this policy will impact our
business.
At the state level, individual states are increasingly aggressive in passing legislation and implementing regulations
designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints,
discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in
some cases, designed to encourage importation from other countries and bulk purchasing. This is increasingly true with
respect to products approved pursuant to the accelerated approval pathway. State Medicaid programs and other payers
are developing strategies and implementing significant coverage barriers, or refusing to cover these products outright,
arguing that accelerated approval drugs have insufficient or limited evidence despite meeting the FDA’s standards for
accelerated approval. In addition, regional healthcare organizations and individual hospitals are increasingly using
bidding procedures to determine what pharmaceutical products and which suppliers will be included in their prescription
drug and other healthcare programs. These measures could reduce the ultimate demand for our Commercial Products
and our product candidates, if approved, or put pressure on our product pricing. We expect that additional state and
federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and
state governments will pay for healthcare products and services, which could result in reduced demand for our
Commercial Products or our product candidates, if approved, or additional pricing pressures.
44
Finally, outside of the U.S., in some countries, including those of the European Union, the pricing of prescription
pharmaceuticals is subject to governmental control and access. In these countries, official list price country pricing
negotiations with governmental authorities can take considerable time after the receipt of marketing approval for a
product. To obtain reimbursement or pricing approval in some countries, we may be required to conduct a clinical trial
that compares the cost-effectiveness of our product to other available therapies.
These measures, as well as others adopted in the future, may result in additional downward pressure on the price that
we receive for our Commercial Products or any of our product candidates, if approved. Accordingly, such reforms, if
enacted, could have an adverse effect on anticipated revenue from our Commercial Products or from product candidates
that we may successfully develop and for which we, or they, may obtain marketing approval and may affect our overall
financial condition and ability to develop or commercialize product candidates.
Our relationships with radiopharmacies, healthcare providers, physicians and third-party payors will be subject to
applicable anti-kickback, fraud and abuse, and other healthcare laws and regulations, which could expose us to
criminal sanctions, civil penalties, contractual damages, reputational harm and diminished profits and future
earnings.
Healthcare professionals, including but not limited to physicians, nurses, medical directors, hospitals, pharmacies,
pharmacy benefit managers, group purchasing organizations, wholesalers, insurers, and all individuals employed by such
entities, which we refer to collectively as HCPs, may influence the recommendation and prescription of our Commercial
Products or any of our product candidates, if approved. Our arrangements with HCPs and others who have the ability to
improperly influence the recommendation and prescription of our Commercial Products or any of our product candidates,
if approved may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may
constrain the business or financial arrangements and relationships through which we market, sell and distribute our
Commercial Products or any of our product candidates, if approved. Restrictions under applicable federal, state and
foreign healthcare laws and regulations include the following:
•the federal healthcare Anti-Kickback Statute prohibits, among other things, persons from knowingly and willfully
soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward
either the referral of an individual for, or the purchase, order, arranging for or recommendation of, any good or
service, for which payment may be made under federal healthcare programs such as Medicare and Medicaid. A
person or entity does not need to have actual knowledge of the federal Anti-Kickback Statute or specific intent to
violate it in order to have committed a violation;
•the FCA imposes criminal and civil penalties, including civil whistleblower or qui tam actions, against individuals or
entities for knowingly presenting or causing to be presented, to the federal government, claims for payment or
approval from Medicare, Medicaid or other government payors that are false or fraudulent or making a false
statement to avoid, decrease or conceal an obligation to pay money to the federal government, with potential
liability including mandatory treble damages and significant per-claim penalties. In addition, the government may
assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute
constitutes a false or fraudulent claim for purposes of the FCA;
•the federal Health Insurance Portability and Accountability Act of 1996 ("HIPAA"), which, in addition to privacy
protections applicable to healthcare providers and other entities, prohibits executing a scheme to defraud any
healthcare benefit program or making false statements relating to healthcare matters. Similar to the federal Anti-
Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to
violate it in order to have committed a violation;
•the federal transparency requirements under the federal Physician Payment Sunshine Act, which requires
manufacturers of drugs, devices, biologics and medical supplies to report to the HHS, information related to
payments and other transfers of value to physicians (as defined by statute), other healthcare providers and
teaching hospitals and ownership and investment interests held by physicians and their immediate family members
and applicable group purchasing organizations;
•analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws, which may
apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-
governmental third-party payors, including private insurers, and certain state laws that require pharmaceutical
companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant
compliance guidance promulgated by the federal government in addition to requiring drug manufacturers to report
information related to payments to physicians and other healthcare providers or marketing expenditures; and
•international, federal or state laws, regulations, or rules that oversee the compounding, administration or
distribution of radiopharmaceutical products by licensed pharmacists.
Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available, it is
possible that some of our business activities could be subject to challenge under one or more of such laws. If our
operations are found to be in violation of any of the laws described above or any other government regulations that apply
to us, we may be subject to penalties, including civil and criminal penalties, damages, fines, exclusion from participation
in government healthcare programs, such as Medicare and Medicaid, imprisonment and the curtailment or restructuring
45
of our operations, any of which could adversely affect our business, financial condition, results of operations and
prospects.
Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and
regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business
practices, including certain advisory agreements we have entered into with physicians who are paid, in part, in the form
of shares or share options, may not comply with current or future statutes, regulations or case law involving applicable
fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these
laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and
administrative penalties, damages, fines, exclusion from government funded healthcare programs, such as Medicare and
Medicaid, and the curtailment or restructuring of our operations. If any of the physicians or other providers or entities
with whom we expect to do business are found to be not in compliance with applicable laws, they may be subject to
criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs. Liabilities
they incur pursuant to these laws could result in significant costs or an interruption in operations, which could have a
material adverse effect on our business, financial condition, results of operations and prospects.
Our reporting and payment obligations under the Medicaid Drug Rebate Program and other governmental drug
pricing programs are complex and may involve subjective decisions. Any failure to comply with those obligations
could subject us to penalties and sanctions.
As a condition of reimbursement by various federal and state health insurance programs, we are required to calculate
and report certain pricing information to federal and state agencies. The regulations governing the calculations, price
reporting and payment obligations are complex and subject to interpretation by various government and regulatory
agencies, as well as the courts. Reasonable assumptions have been made where there is lack of regulations or clear
guidance and such assumptions involve subjective decisions and estimates. We are required to report any revisions to
our calculation, price reporting and payment obligations previously reported or paid. Such revisions could affect our
liability to federal and state payors and also adversely impact our reported financial results of operations in the period of
such restatement. Further, a number of states have either implemented or are considering implementation of drug price
transparency legislation that may prevent or limit our ability to take price increases at certain rates or frequencies.
Requirements under such laws include advance notice of planned price increases, reporting price increase amounts and
factors considered in taking such increases, wholesale acquisition cost information disclosure to prescribers, purchasers,
and state agencies, and new product notice and reporting. Such legislation could limit the price or payment for certain
drugs, and a number of states are authorized to impose civil monetary penalties or pursue other enforcement
mechanisms against manufacturers for the untimely, inaccurate, or incomplete reporting of drug pricing information or for
otherwise failing to comply with drug price transparency requirements. If we are found to have violated state law
requirements, we may become subject to significant penalties or other enforcement mechanisms, which could have a
material adverse effect on our business.
Uncertainty exists as new laws, regulations, judicial decisions, or new interpretations of existing laws, or regulations
related to our calculations, price reporting or payments obligations increases the chances of a legal challenge,
restatement or investigation. If we become subject to investigations, restatements, or other inquiries concerning our
compliance with price reporting laws and regulations, we could be required to pay or be subject to additional
reimbursements, penalties, sanctions or fines, which could have a material adverse effect on our business, financial
condition and results of operations. In addition, it is possible that future healthcare reform measures could be adopted,
which could result in increased pressure on pricing and reimbursement of our Commercial Products and any of our
product candidates, if approved, and thus have an adverse impact on our financial position or business operations.
Further, state Medicaid programs may be slow to invoice pharmaceutical companies for calculated rebates resulting in a
lag between the time a sale is recorded and the time the rebate is paid. This results in us having to carry a liability on our
consolidated balance sheets for the estimate of rebate claims expected for Medicaid patients. If actual claims are higher
than current estimates, our financial position and results of operations could be adversely affected.
In addition to retroactive rebates and the potential for 340B Program refunds, if we are found to have knowingly
submitted any false price information related to the Medicaid Drug Rebate Program to CMS, we may be liable for civil
monetary penalties. Such failure could also be grounds for CMS to terminate our Medicaid drug rebate agreement,
pursuant to which we participate in the Medicaid program. In the event that CMS terminates our rebate agreement,
federal payments may not be available under government programs, including Medicaid or Medicare Part B, for our
covered outpatient drugs.
Additionally, if we overcharge the government in connection with the Federal Supply Schedule pricing program or Tricare
Retail Pharmacy Program, whether due to a misstated Federal Ceiling Price or otherwise, we are required to refund the
difference to the government. Failure to make necessary disclosures and/or to identify contract overcharges can result in
allegations against us under the FCA and other laws and regulations. Unexpected refunds to the government, and
responding to a government investigation or enforcement action, would be expensive and time-consuming, and could
have a material adverse effect on our business, financial condition, results of operations and growth prospects.
Our collaborators are also subject to similar requirements outside of the U.S. and thus the attendant risks and
uncertainties. If our collaborators suffer material and adverse effects from such risks and uncertainties, our rights and
benefits for our licensed products could be negatively impacted, which could have a material and adverse impact on our
revenues.
46
We are subject to stringent privacy laws, information security laws, regulations, policies and contractual obligations
related to data privacy and security and changes in such laws, regulations, policies, contractual obligations and
failure to comply with such requirements could subject us to significant fines and penalties, which may have a
material adverse effect on our business, financial condition or results of operations.
We are subject to data privacy and protection laws and regulations that apply to the collection, transmission, storage and
use of personal information, which among other things, impose certain requirements relating to the privacy, security and
transmission of personal information, including comprehensive regulatory systems in the U.S., European Union, United
Kingdom, Australia, and other countries in which we may conduct business. The legislative and regulatory landscape for
privacy and data protection continues to evolve in jurisdictions worldwide, and there has been an increasing focus on
privacy and data protection issues with the potential to affect our business. Failure to comply with any of these laws and
regulations could result in enforcement action against us, including fines, imprisonment of company officials and public
censure, claims for damages by affected individuals, damage to our reputation and loss of goodwill, any of which could
have a material adverse effect on our business, financial condition, results of operations or prospects.
There are numerous U.S. federal and state laws and regulations related to the privacy and security of personal
information. In particular, regulations promulgated pursuant to HIPAA, establish privacy and security standards that limit
the use and disclosure of protected health information and require the implementation of administrative, physical and
technological safeguards to protect the privacy of protected health information and ensure the confidentiality, integrity
and availability of electronic protected health information. Determining whether protected health information has been
handled in compliance with applicable privacy standards and our contractual obligations can be complex and may be
subject to changing interpretation.
If we fail to comply with applicable privacy laws, including applicable HIPAA privacy and security standards, we could
face civil and criminal penalties. HHS enforcement activity can result in financial liability and reputational harm, and
responses to such enforcement activity can consume significant internal resources. HHS's Office for Civil Rights ("OCR")
has been especially active in investigating and enforcing actual or alleged non-compliance with the HIPAA rules. In
addition, state attorneys general are authorized to bring civil actions seeking either injunctions or damages in response
to violations that threaten the privacy of state residents. Some new state laws, such as the state of Washington's My
Health My Data Act, include a private right of action. We cannot be sure how these regulations will be interpreted,
enforced or applied to our operations. In addition to the risks associated with enforcement activities and potential
contractual liabilities, our ongoing efforts to comply with evolving laws and regulations at the federal and state level may
be costly and require ongoing modifications to our policies, procedures and systems.
In addition to potential enforcement by the HHS, we could also be potentially subject to privacy enforcement from the
FTC. The FTC has been particularly focused on the unpermitted processing of health and genetic data through its recent
enforcement actions and is expanding the types of privacy violations that it interprets to be “unfair” under Section 5 of
the FTC Act, as well as the types of activities it views to trigger the Health Breach Notification Rule (which the FTC also
has the authority to enforce). The agency is also in the process of developing rules related to commercial surveillance
and data security. We will need to account for the FTC’s evolving rules and guidance for proper privacy and data security
practices in order to mitigate risk for a potential enforcement action, which may be costly. Finally, both the FTC and
HHS’s enforcement priorities (as well as those of other federal regulators) may be impacted by the change in
administration and new leadership. These shifts in enforcement priorities may also impact our business.
There are also increased restrictions at the federal level relating to transferring sensitive data outside of the U.S. to
certain foreign countries. The Protecting Americans’ Data from Foreign Adversaries Act of 2024. This law creates certain
restrictions for entities that disclose sensitive data (including potential health data) to countries such as China. Failure to
comply with these rules can lead to a potential FTC enforcement action. Additionally, the Department of Justice recently
finalized a rule which creates similar restrictions related to the transfer of sensitive U.S. data to countries such as China.
These data transfer restrictions (and others that may pass in the future) may create operational challenges and legal
risks for our business.
States are also active in creating specific rules relating to the processing of personal information. In 2018, California
passed into law the California Consumer Privacy Act ("CCPA"), which took effect on January 1, 2020 and imposed many
requirements on businesses that process the personal information of California residents. Many of the CCPA’s
requirements are similar to those found in the European Union's General Data Protection Regulation ("GDPR"), which is
further described below, including requiring businesses to provide notice to data subjects regarding the information
collected about them and how such information is used and shared, including use in marketing, and providing data
subjects the right to request access to such personal information and, in certain cases, request the erasure of such
personal information. The CCPA also affords California residents the right to opt-out of “sales” of "sharing" of their
personal information under certain circumstances. The CCPA contains significant penalties for companies that violate its
requirements.
In November 2020, California voters passed a ballot initiative for the California Privacy Rights Act ("CPRA"), which went
into effect on January 1, 2023 and significantly expanded the CCPA to incorporate additional GDPR-like provisions
including requiring that the use, retention and sharing of personal information of California residents be reasonably
necessary and proportionate to the purposes of collection or processing, granting additional protections for sensitive
personal information, and requiring greater disclosures related to notice to residents regarding retention of information.
The CPRA also created a new enforcement agency – the California Privacy Protection Agency or CPPA – the sole
responsibility of which is to enforce the CPRA and other California privacy laws, which will further increase compliance
47
risk. The CCPA also has rulemaking authority under the CCRA with recent regulations finalized for, among other things,
cybersecurity audits, risk assessment, and use of automated decision-making technologies. The provisions in the CPRA
may apply to some of our business activities. In addition to California, at least 19 other states have passed
comprehensive privacy laws. These laws may impact our business activities if and to the extent applicable to our
business, including our identification of research subjects, relationships with business partners, and ultimately the
marketing and distribution of our Commercial Products and our product candidates, if approved.
Plaintiffs’ lawyers are also increasingly using privacy-related statutes at both the state and federal level to bring lawsuits
against companies for their data-related practices. In particular, there have been a significant number of cases filed
against companies for their use of pixels and other tracking technologies, chatbots, and session replay technologies.
These cases often allege violations of the California Invasion of Privacy Act and other state laws regulating wiretapping,
as well as the federal Video Privacy Protection Act and Electronic Communications Protection Act. The rise in these types
of lawsuits creates potential risk for our business. Additionally, privacy breach class actions under various U.S. state laws
remain highly active which could result in potential litigation exposure in the event that we experience a privacy breach.
Similar to the laws in the U.S., there are significant privacy and data security laws that apply in Europe, the United
Kingdom, and other countries. The collection, use, disclosure, transfer, or other processing of personal data, including
personal health data, regarding individuals who are located in the EEA, and the processing of personal data that takes
place in the EEA, is regulated by the GDPR, which went into effect in May 2018 and which imposes obligations on
companies that operate in our industry with respect to the processing of personal data and the cross-border transfer of
such data. The GDPR imposes onerous accountability obligations requiring data controllers and processors to maintain a
record of their data processing and policies. If our or our partners’ or service providers’ privacy or data security measures
fail to comply with the GDPR requirements, we may be subject to litigation, regulatory investigations, enforcement
notices requiring us to change the way we use personal data and/or fines of up to 20 million Euros or up to 4% of the
total worldwide annual turnover of the group of companies of the preceding financial year, whichever is higher, as well as
compensation claims by affected individuals, negative publicity, reputational harm and a potential loss of business and
goodwill.
Certain privacy and data protection laws, including the GDPR, restrict the cross-border transfer of personal data from the
original jurisdiction to countries that have not been found to offer "adequate protection" for personal data. Privacy
advocates have successfully challenged prior compliance mechanisms, such as the EU-U.S. Privacy Shield, prompting
ongoing concerns about the ability of companies to transfer personal data from the European Union to other countries.
In October 2022, President Biden signed an executive order to implement the EU-U.S. Data Privacy Framework, which
serves as a replacement to the EU-U.S. Privacy Shield. An adequacy decision, adopted by the European Commission on
July 10, 2023 permits U.S. companies who self-certify to the EU-U.S. Data Privacy Framework to rely on it as a valid data
transfer mechanism for data transfers from the European Union to the U.S. To date, the EU-U.S. Data Privacy Framework
has withstood challenges from privacy advocates, including in a September 2025 decision by the European General
Court upholding the validity of the European Commission's 2023 adequacy decision in Latombe v. Commission. However,
if any subsequent challenges are successful, they may not only impact the EU-U.S. Data Privacy Framework, but also
further limit the viability of the standard contractual clauses and other data transfer mechanisms. The uncertainty around
this issue has the potential to impact our business. Following the withdrawal of the United Kingdom from the European
Union, the United Kingdom Data Protection Act 2018 ("UK Data Protection Act") applies to the processing of personal
data that takes place in the United Kingdom and includes parallel obligations to those set forth by GDPR. In relation to
data transfers, both the United Kingdom and the European Union have determined, through separate “adequacy”
decisions, that data transfers between the two jurisdictions are in compliance with the UK Data Protection Act and the
GDPR, respectively. The United Kingdom and the U.S. have also agreed to a U.S.-UK “Data Bridge”, which functions
similarly to the EU-U.S. Data Privacy Framework and provides an additional legal mechanism for companies to transfer
data from the United Kingdom to the U.S.
Switzerland has also approved an adequacy decision in relation to the Swiss-U.S. Data Privacy Framework (which
functions similarly to the EU-U.S. Data Privacy Framework and the U.S.-UK Data Bridge in relation to data transfers from
Switzerland to the U.S.). Any changes or updates to these developments have the potential to impact our business.
Beyond GDPR, there are privacy and data security laws in a growing number of countries around the world. While many
loosely follow GDPR as a model, other laws contain different or conflicting provisions. These laws will impact our ability to
conduct our business activities, including both our clinical trials and the sale and distribution of Commercial Products,
through increased compliance costs, costs associated with contracting and potential enforcement actions.
In Australia, we are subject to the Privacy Act 1988 (Cth) and the Australian Privacy Principles ("APPs"), which regulate
the collection, use, disclosure, storage and security of personal information, including health information, which is treated
as sensitive information subject to heightened protections. The Privacy Legislation Amendment (Enforcement and Other
Measures) Act 2022 significantly increased maximum penalties for serious or repeated interferences with privacy to the
greater of A$50 million, three times the value of any benefit obtained from the breach, or 30% of adjusted turnover in the
relevant period. We are also subject to the Notifiable Data Breaches scheme, which requires entities to notify affected
individuals and the Office of the Australian Information Commissioner ("OAIC"), of eligible data breaches that are likely to
result in serious harm. The Australian government has signaled further reforms to privacy legislation, including proposals
to strengthen individual rights, expand the definition of personal information, and introduce a statutory tort for serious
invasions of privacy. If enacted, such reforms could impose additional compliance obligations and increase our exposure
to regulatory enforcement and private claims. Failure to comply with Australian privacy requirements could result in
48
significant penalties, reputational harm, regulatory investigations and remedial orders, any of which could have a material
adverse effect on our business, financial condition or results of operations.
While we continue to address the implications of the recent changes to data privacy regulations, data privacy remains an
evolving landscape at both the domestic and international level, with new regulations coming into effect and continued
legal challenges, and our efforts to comply with the evolving data protection rules may be unsuccessful. It is possible
that these laws may be interpreted and applied in a manner that is inconsistent with our practices. We must devote
significant resources to understanding and complying with this changing landscape. Failure to comply with laws
regarding data protection would expose us to risk of enforcement actions taken by data protection authorities in the EEA
and Australia and elsewhere and carries with it the potential for significant penalties if we are found to be non-compliant.
Similarly, failure to comply with federal and state laws in the U.S. regarding privacy and security of personal information
could expose us to penalties under such laws. Any such failure to comply with data protection and privacy laws could
result in government-imposed fines or orders requiring that we change our practices, claims for damages or other
liabilities, regulatory investigations and enforcement action, litigation and significant costs for remediation, any of which
could adversely affect our business. Even if we are not determined to have violated these laws, government
investigations into these issues typically require the expenditure of significant resources and generate negative publicity,
which could harm our business, financial condition, results of operations or prospects.
If we fail to comply with environmental, health and safety laws and regulations, including those governing
radiopharmaceutical products and radioactive materials, we could become subject to fines or penalties or incur costs
that could have a material adverse effect on our business.
We are subject to numerous environmental, health and safety laws and radiation safety regulations, including those
governing laboratory procedures and the handling, use, storage, treatment, transportation and disposal of hazardous
materials and wastes. Our operations involve the use, storage, treatment, and transportation of hazardous and flammable
materials, including chemicals and biological and radioactive materials. While most of the activities are conducted by
third-party partners on our behalf or by pharmacists or healthcare professionals consistent with their own professional
obligations on their own behalf, our operations also produce hazardous waste products and allow the decay of
radioactive material (where legally allowed) on site prior to disposal as non-radioactive waste. We generally contract with
third parties for the disposal of these materials and wastes. We cannot eliminate the risk of contamination or injury from
these materials. In the event of contamination or injury resulting from our use of hazardous materials, we could be held
liable for any resulting damages, and any liability could exceed our resources. We also could incur significant costs
associated with civil or criminal fines and penalties.
Transportation of radioactive, biological, and/or hazardous materials is highly regulated by each governmental
jurisdiction we operate in. Additionally, the physical transportation of these materials puts us at risk of vehicular
accidents which could result in the loss of control of these materials and potential environmental and human health risks
and related regulatory actions against us.
Our use of facilities that use and produce radioactive materials subjects us to compliance with D&D requirements when
we close those facilities, exposing us to potentially significant costs. Our product candidates are manufactured using
radioactive components. When a cyclotron reaches the end of its useful life at one of our facilities or if we need to
abandon such facility for any other reason, we are obligated under the laws and regulatory rules of the various
jurisdictions in which we operate to decommission and decontaminate such facility or cyclotron. Estimating the amount
and timing of such future D&D costs includes, among other factors, country-specific requirements and projections as to
when a facility will retire or the useful life of a cyclotron. If we do not conduct D&D properly at any of our sites, we may
suffer significant additional costs to remediate any D&D deficiencies, fines, regulatory or criminal charges or other
sanction or legal action, any of which could have a material adverse effect upon our business, financial condition and
results of operations. Although we have estimated our future D&D costs and recorded a liability for such costs, there can
be no assurances that we will not incur material D&D costs beyond such estimates or our provisions.
Although we maintain workers’ compensation insurance to cover us for costs and expenses we may incur due to injuries
to our employees resulting from the use of hazardous materials, this insurance may not provide adequate coverage
against potential liabilities. We do not maintain insurance for environmental liability or toxic tort claims that may be
asserted against us in connection with our storage or disposal of biological, hazardous or radioactive materials.
In addition, we may incur substantial costs in order to comply with current or future environmental, health and safety
laws and regulations. These current or future laws and regulations may impair our research, development or
commercialization efforts. Failure to comply with these laws and regulations also may result in substantial fines, penalties
or other sanctions.
The use of hazardous materials, including radioactive and biological materials, in our research and development
efforts imposes certain compliance costs on us and may subject us to liability for claims arising from the use or
misuse of these materials.
Our research, development and manufacturing activities involve the controlled use of hazardous materials, including
chemicals, radioactive and biological materials, such as radioisotopes. We and our third-party manufacturers are subject
to federal, state, local and foreign environmental laws and regulations governing, among other matters, the handling,
storage, use, transportation and disposal of these materials and some waste products.
49
Our use of chemicals in the manufacturing process for our Commercial Product and product candidates is also subject to
chemicals approvals, registrations and regulations around the world, including a regulation in the European Union known
as Registration, Evaluation, Authorization and Restriction of Chemicals, and similar laws and regulations in certain other
jurisdictions in which we operate. In addition, we are required to obtain and maintain a hazardous materials license,
pursuant to which we are required to perform annual self-audits, and that may result in random inspections by regulators.
If such audit or inspection were to result in adverse findings, it may impact our ability to maintain our license, which
would in turn adversely affect our ability to conduct our business.
Additionally, we cannot completely eliminate the risk of contamination or injury from these materials, and we could be
held liable for any damages that result, which could exceed our financial resources. We currently maintain insurance
coverage for injuries resulting from the hazardous materials we use; however, future claims may exceed the amount of
our coverage. Also, we do not have insurance coverage for pollution cleanup and removal. Currently the costs of
complying with such federal, state, local and foreign environmental regulations are not significant, and consist primarily
of waste disposal expenses. However, they could become expensive, and current or future environmental laws or
regulations may impair our research, development, production and commercialization efforts.
Although we intend to validate that any third-party manufacturers’ procedures for using, handling, storing and disposing
of these materials comply with legally prescribed standards, we cannot completely eliminate the risk of contamination or
injury resulting from medical or hazardous materials. As a result of any such contamination or injury, we may incur liability
or local, city, state or federal authorities may curtail the use of these materials and interrupt our business operations. In
the event of an accident, we could be held liable for damages or penalized with fines, and the liability could exceed our
resources. Comparable restrictions and related risks regarding the use of potentially hazardous substances are also
applicable outside the U.S. Compliance with applicable environmental laws and regulations is expensive, and current or
future environmental regulations may impair our research, development and production efforts, which could harm our
business, financial condition, results of operations and prospects.
Laws and regulations governing international operations may preclude us from developing, manufacturing and
selling certain products outside of the U.S. and require us to develop and implement costly compliance programs.
We are subject to numerous laws and regulations in each jurisdiction outside of the U.S. in which we operate. The
creation, implementation and maintenance of international business practices compliance programs is costly and such
programs are difficult to enforce, particularly where reliance on third parties is required.
The U.S. Foreign Corrupt Practices Act ("FCPA") prohibits any U.S. individual or business from paying, offering,
authorizing payment or offering of anything of value, directly or indirectly, to any foreign official, political party or
candidate for the purpose of influencing any act or decision of the foreign entity in order to assist the individual or
business in obtaining or retaining business. The FCPA also obligates companies whose securities are listed in the U.S. to
comply with certain accounting provisions requiring us to maintain books and records that accurately and fairly reflect all
transactions of the corporation, including international subsidiaries, and to devise and maintain an adequate system of
internal accounting controls. The FCPA is enforced by the DOJ and the SEC.
Compliance with the FCPA is expensive and difficult, particularly in countries in which corruption is a recognized problem.
In addition, the FCPA presents particular challenges in the pharmaceutical industry, because, in many countries,
hospitals, clinics, universities and similar institutions are operated by the government, and doctors and other healthcare
professionals are considered foreign officials. Certain payments to healthcare professionals in connection with clinical
trials, regulatory approvals, sales and marketing, and other work have been deemed to be improper payments to
government officials and have led to FCPA enforcement actions. Because the FCPA applies to indirect payments, the use
of third parties and other collaborators can increase potential FCPA risk, as we could be held liable for the acts of third
parties that do not comply with the FCPA’s requirements.
The failure to comply with laws governing international business practices may result in substantial penalties, including
suspension or debarment from government contracting. Violation of the FCPA can result in significant civil and criminal
penalties. Indictment alone under the FCPA can lead to suspension of the right to do business with the U.S. government
until the pending claims are resolved. Conviction of a violation of the FCPA can result in long-term disqualification as a
government contractor. The termination of a government contract or relationship as a result of our failure to satisfy any
of our obligations under laws governing international business practices would have a negative impact on our operations
and harm our reputation and ability to procure government contracts. The SEC also may suspend or bar issuers from
trading securities on U.S. exchanges for violations of the FCPA’s accounting provisions.
Like the FCPA, the Australian Criminal Code, the U.K. Bribery Act and other anti-corruption laws throughout the world
similarly prohibit offers and payments made to obtain improper business advantages, including offers or payments to
healthcare professionals and other government and non-government officials. These other anti-corruption laws also can
result in substantial financial penalties and other collateral consequences.
Various laws, regulations and executive orders also restrict the use and dissemination outside of the U.S., or the sharing
with certain non-U.S. nationals, of information classified for national security purposes, as well as certain products and
technical data relating to those products. Our expansion outside of the U.S., has required, and will continue to require, us
to dedicate additional resources to comply with these laws, and these laws may preclude us from developing,
manufacturing, or selling certain drugs and product candidates, if approved, outside of the U.S., which could limit our
growth potential and increase our development costs.
50
Changes in U.S. and international trade policies may adversely impact our business and operating results.
The U.S. government has made statements and taken certain actions that have led to significant changes to U.S. and
international trade policies, including imposing several rounds of tariffs and export control restrictions affecting products
manufactured in certain foreign jurisdictions. Although trade policies continue to evolve, there is ongoing uncertainty as
to whether there will be further changes. It is unknown whether and to what extent new tariffs, export controls, or other
new laws or regulations will be adopted, or the effect that any such actions would have on us or our industry.
Further, some of our manufacturers and suppliers are located outside the United States, including in China. We are
exposed to the possibility of product supply disruption and increased costs and expenses in the event of changes to the
laws, rules, regulations and policies of the governments of countries in which we or our suppliers operate. Certain foreign
biotechnology companies may become subject to trade restrictions, sanctions, other regulatory requirements or
legislation by the U.S. government, which could restrict or even prohibit our ability to work with such entities, thereby
potentially disrupting their supply of material to us. Such disruptions could have adverse effects on the development of
our product candidates and our business operations.
For example, the pharmaceutical industry generally, and in some instances our Company or our collaborators or other
third parties on which we rely, depend on China-based suppliers or service providers for certain raw materials, products
and services, or other activities. Our ability or the ability of our collaborators or such other third parties to continue to
engage these China-based suppliers or service providers for certain preclinical research programs and clinical
development programs could be restricted due to geopolitical developments between the U.S. and China, including as a
result of the escalation of tariffs or other trade restrictions or the recently enacted BIOSECURE Act. In any such
circumstances, we may not be able to engage a backup or alternative supplier or service provider in a timely manner or
at all. This, in turn, could materially and adversely affect our or our collaborators' ability to manufacture or supply
marketed products and product candidates or advance our or our collaborators' preclinical research or clinical
development programs, which could materially and adversely affect our business and future prospects.
Any unfavorable government policies on international trade, such as export controls, capital controls or tariffs, may
increase the cost of manufacturing our Commercial Products and product candidates and platform materials, affect the
demand for our Commercial Products and product candidates, if approved, the competitive position of our Commercial
Products and product candidates, if approved, and import or export of raw materials and finished Commercial Products
and product candidates used in our and our collaborators’ preclinical studies and clinical trials, particularly with respect
to any materials that we import from China. If any new tariffs, export controls, legislation and/or regulations are
implemented, or if existing trade agreements are renegotiated or, in particular, if either the U.S. or Chinese government
takes retaliatory trade actions due to the recent trade tension, such changes could have an adverse effect on our
business, financial condition and results of operations.
With the passage of the CREATES Act, we are exposed to possible litigation and damages by competitors who may
claim that we are not providing sufficient quantities of our approved products on commercially reasonable, market-
based terms for testing in support of their abbreviated new drug applications ("ANDAs"), 505(b)(2) NDAs and
biosimilar product applications.
In December 2019, President Trump signed legislation intended to facilitate the development of generic and biosimilar
products. The bill, previously known as the CREATES Act, authorizes sponsors of ANDAs, 505(b)(2) NDAs, or biosimilar
product applications to file lawsuits against companies holding NDAs or BLAs that decline to provide sufficient quantities
of an approved reference drug or biological product on commercially reasonable, market-based terms. Drug or biological
products on FDA’s drug shortage list are exempt from these new provisions unless the product has been on the list for
more than six continuous months or the FDA determines that the supply of the product will help alleviate or prevent a
shortage.
To bring an action under the statute, the developer of a product candidate that seeks to develop the product and seek
approval under an ANDA, 505(b)(2) NDA, or biosimilar product application must take certain steps to request the
reference product from the reference product manufacturer, which, in the case of products covered by a REMS with
elements to assure safe use, include obtaining authorization from the FDA for the acquisition of the reference product. If
the reference product manufacturer does not provide the reference product and the ANDA, 505(b)(2) NDA, or biosimilar
product sponsor does bring an action for failure to provide a reference product, there are certain affirmative defenses
available to the reference product manufacturer, which must be shown by a preponderance of evidence, including that
the NDA or BLA holder sells the reference product through agents, distributors, or wholesalers and has placed no
restrictions, explicit or implicit, on selling the reference product to ANDA, 505(b)(2) or biosimilar sponsors. If the sponsor
prevails in litigation, it is entitled to a court order directing the reference product manufacturer to provide, without delay,
sufficient quantities of the applicable product on commercially reasonable, market-based terms, plus reasonable
attorney fees and costs.
Additionally, the new statutory provisions authorize a federal court to award the product developer an amount “sufficient
to deter” the reference product manufacturer from refusing to provide sufficient product quantities on commercially
reasonable, market-based terms, up to a certain maximum amount based on revenue earned while in noncompliance, if
the court finds, by a preponderance of the evidence, that the reference product manufacturer did not have a legitimate
business justification to delay providing the product or failed to comply with the court’s order. For the purposes of the
statute, the term “commercially reasonable, market-based terms” is defined as (i) the nondiscriminatory price at or below
51
the most recent wholesale acquisition cost for the product, (ii) a delivery schedule that meets the statutorily defined
timetable, and (iii) no additional conditions on the sale.
Although we intend to comply fully with the terms of these statutory provisions, we are still exposed to potential litigation
and damages by competitors who may claim that we are not providing sufficient quantities of our Commercial Products
on commercially reasonable, market-based terms for testing in support of ANDAs, 505(b)(2) NDA applications or
biosimilar product applications. Such litigation would subject us to additional litigation costs, damages and reputational
harm, which could lead to lower revenues. The CREATES Act may facilitate future competition with our Commercial
Products and any of our product candidates, if approved, which could impact our ability to maximize product revenue.
We are required to comply with governmental economic and trade sanctions and export and import controls that
could impair our or our collaborators’ ability to compete in international markets due to licensing requirements and
subject us or them to liability if we or they are not in compliance with applicable laws.
Our Commercial Products and product candidates are subject to international, national and state export control and
import laws and regulations, including the U.S. Export Administration Regulations, U.S. Customs regulations, and we are
required to comply with these laws as well as various economic and trade sanctions, including those administered by the
U.S. Treasury Department’s Office of Foreign Assets Controls. These laws and regulations restrict our ability to transact
or deal with certain countries, regions, governments, persons and entities. Our activities, including our procurement of
materials and exports of our Commercial Products and product candidates, must be in compliance with these laws and
regulations. While we have policies and procedures designed to ensure that we maintain compliance with these laws and
regulations, there is a risk that our employees, agents, or business partners may take actions in violation of our policies
and applicable law, for which we may be ultimately held responsible.
If we fail to comply with these laws and regulations, we and certain of our employees could be subject to substantial civil
or criminal penalties, including the possible loss of export or import privileges; fines, which may be imposed on us or our
collaborators and the respective responsible employees or managers; and, in extreme cases, the incarceration of
responsible employees or managers. Investigations of alleged violations can be expensive and disruptive, and such
violation (or allegation of a violation) could materially adversely affect our reputation, business, financial condition and
results of operations.
In addition, changes in our Commercial Products or changes in applicable export or import laws and regulations may
create delays in the introduction, provision, or sale of our Commercial Products in international markets, prevent
customers from using our Commercial Products or, in some cases, prevent the export or import of our Commercial
Products to certain countries, governments or persons altogether. Any limitation on our ability to export, provide, or sell
our Commercial Products could adversely affect our business, financial condition and results of operations.
Risks Related to Our Dependence on Third Parties
We depend on collaborations with third parties for certain aspects of the development, marketing and/or
commercialization of Commercial Products and our product candidates, if approved. If those collaborations are not
successful, or if we are not able to maintain our existing collaborations or establish additional collaborations, we may
have to alter our development and commercialization plans and may not be able to capitalize on the market potential
of Commercial Products or our product candidates, if approved.
Our product development programs and the commercialization of our Commercial Products and product candidates, if
approved, require local expertise and substantial additional cash to fund expenses. We expect to maintain our existing
collaborations and collaborate with additional pharmaceutical and biotechnology companies for certain aspects of the
development, marketing and/or commercialization of our Commercial Products and product candidates. For example, we
expect to rely on additional partners to develop and commercialize our Commercial Products outside of the U.S.,
including our ongoing partnership with Grand Pharmaceutical Group Limited ("Grand Pharma") for our imaging and
therapeutic product candidates in Greater China. In addition, we intend to utilize collaborators to aid in the further
development, potential marketing and/or commercialization of our product candidates. We also have a license agreement
with Eli Lilly and Company Ltd ("Lilly") for the exclusive worldwide rights to develop and commercialize radiolabeled
forms of olaratumab together with our linker and our other proprietary licensed technology, for the diagnosis and
treatment of human cancers.
Potential collaborators include large and mid-size pharmaceutical companies, regional and national pharmaceutical
companies and biotechnology companies and we face significant competition in seeking appropriate collaborators,
including as a result of a significant number of recent business combinations among large pharmaceutical companies that
have reduced the number of potential collaborators. Whether we reach a definitive agreement for a collaboration will
depend, among other things, upon the assessment of the potential collaborator’s expertise, its current and expected
resources and competing priorities, the terms and conditions of the proposed collaboration and the proposed
collaborator’s evaluation of a number of factors. Those factors may include the design or results of clinical trials, the
likelihood of approval by the FDA or foreign regulatory authorities, the potential market for the product or product
candidate, the costs and complexities of manufacturing and delivering such product or product candidate, if approved, to
patients, the potential of competing products, the existence of uncertainty with respect to our ownership of intellectual
property, which can exist if there is a challenge to such ownership without regard to the merits of the challenge, and
industry and market conditions generally. A potential collaborator may also consider alternative product candidates or
52
technologies for similar indications that may be available to collaborate on and whether such a collaboration could be
more attractive than the one with us.
Collaborations are complex and time-consuming to negotiate, document and manage. We may not be able to negotiate
collaborations on a timely basis, on acceptable terms, or at all, or we may be restricted under then-existing collaboration
agreements from entering into future agreements on certain terms with potential collaborators. If we are unable to
maintain our current collaboration agreements or enter into new collaboration agreements, we may have to curtail,
reduce or delay the development or commercialization programs for our Commercial Products or product candidates, if
approved, or increase our expenditures and undertake development or commercialization activities at our own expense.
If we elect to increase our expenditures to fund and undertake development or commercialization activities on our own,
we may need to obtain additional expertise and additional capital, which may not be available to us on acceptable terms,
or at all. If we do not have sufficient funds or expertise to undertake the necessary development and commercialization
activities, we may not be able to further develop our Commercial Products or product candidates or bring them to market
and generate product revenue, if approved.
Our ability to generate revenues from these arrangements will depend on our collaborators’ abilities to successfully
perform the functions assigned to them in these arrangements, and our collaboration agreements may not lead to the
development or commercialization of future products or our product candidates, if approved, in the most efficient
manner, or at all, and may result in lower product revenues or profitability to us than if we were to market and sell our
Commercial Products or our product candidates, if approved, ourselves. In connection with any such arrangements with
third parties, we will likely have limited control over the amount and timing of resources that our collaborators dedicate to
the development, marketing and/or commercialization of our Commercial Products or product candidates. Further, if our
collaborations do not result in the successful development and commercialization of our future products or product
candidates, if approved, or if any one of our collaborators terminates its agreement with us, we may not receive any
future milestone or royalty payments under the collaboration. If we do not receive the funding we expect under these
agreements, the development and commercialization of future products or product candidates, if approved, could be
delayed and we may need additional resources to develop product candidates.
Collaborations involving our Commercial Products and product candidates pose the following risks to us:
•collaborators have significant discretion in determining the efforts and resources that they will apply to these
collaborations;
•collaborators may not perform their obligations as expected or in compliance with applicable local and national laws
and regulatory requirements;
•collaborators may de-emphasize or may not pursue development, marketing and/or commercialization of our
Commercial Products or product candidates or may elect not to continue or renew development, marketing or
commercialization programs based on clinical trial results, changes in the collaborator’s strategic focus, including as
a result of a sale or disposition of a business unit or development function, or available funding or external factors
such as an acquisition that diverts resources or creates competing priorities;
•collaborators may delay clinical trials, provide insufficient funding for a clinical trial program, stop a clinical trial or
abandon a product candidate, repeat or conduct new clinical trials or require a new formulation of a product
candidate for clinical testing;
•collaborators could independently develop, or develop with third parties, products that compete directly or
indirectly with our Commercial Products or product candidates if the collaborators believe that competitive
products are more likely to be successfully developed or can be commercialized under terms that are more
economically attractive than ours;
•a collaborator with marketing and distribution rights to one or more products or product candidates may not commit
sufficient resources to the marketing and distribution of our Commercial Products or product candidates;
•disagreements with collaborators, including disagreements over proprietary rights, contract interpretation or the
preferred course of development or commercialization, might cause delays or termination of the research,
development or commercialization of products or product candidates, might lead to additional responsibilities for us
with respect to our Commercial Products or product candidates, or might result in litigation or arbitration, any of
which would be time-consuming and expensive;
•collaborators may not properly maintain or defend our intellectual property rights or may use our proprietary
information in such a way as to invite litigation that could jeopardize or invalidate our intellectual property or
proprietary information or expose us to potential litigation;
•collaborators may infringe the intellectual property rights of third parties, which may expose us to litigation and
potential liability;
•we may lose certain valuable rights under circumstances identified in any collaboration arrangement that we enter
into, such as if we undergo a change of control;
53
•collaborations may be terminated and, if terminated, may result in a need for additional capital to pursue further
development, marketing and/or commercialization of the applicable products or product candidates or to enter into
new collaboration agreements;
•collaborators may learn about our discoveries and use this knowledge to compete with us in the future;
•collaboration agreements may not lead to development or commercialization of product candidates in the most
efficient manner or at all; and
•the number and type of our collaborations could adversely affect our attractiveness to other collaborators or
acquirers.
If any of these events occurs, the market potential of our Commercial Products and product candidates, if approved,
could be reduced, and our business could be materially harmed.
If we are unable to establish and maintain our agreements with third parties to distribute our Commercial Products to
patients, our results of operations and business could be adversely affected.
We rely substantially on third parties to commercially distribute our Commercial Products to patients. For example, we
have contracted with a distribution network of specialty pharmacies, which sell our Commercial Products directly to
patients, and specialty distributors, which sell our Commercial Products to healthcare entities who then resell to patients.
While we have entered into agreements with each of these pharmacies and distributors to distribute our Commercial
Products in the U.S., they may not perform as agreed or they may terminate their agreements with us. We may also need
to enter into agreements with additional pharmacies or distributors, and there is no guarantee that we will be able to do
so on a timely basis, at commercially reasonable terms, or at all. If we are unable to maintain and, if needed, expand, our
network of specialty pharmacies and specialty distributors, we would be exposed to substantial distribution risk. In
addition, and particularly as we expand into less-mature markets or into countries where corruption may be more
prevalent, we will need to conduct robust due diligence with third-party collaboration partners to best ensure that our
Commercial Products and our other product candidates, if approved, are able to be manufactured, compounded, or
distributed on a timely basis that complies will all applicable laws, regulations, and rules, including but not limited to,
those that deal with anti-corruption, anti-kickback, marketing authorization and distribution of pharmaceutical products,
the environment, and the safe use of the products with patients.
The use of specialty pharmacies and specialty distributors involves certain risks, including, but not limited to, risks that
these organizations will:
•not provide us accurate or timely information regarding their inventories, the number of patients who are using our
Commercial Products or serious adverse reactions, events and/or product complaints regarding our Commercial
Products;
•not effectively sell or support our Commercial Products or communicate publicly concerning our Commercial
Products in a manner that is contrary to FDA rules and regulations;
•reduce their efforts or discontinue to sell or support, or otherwise not effectively sell or support, our Commercial
Products;
•not devote the resources necessary to sell our Commercial Products in the volumes and within the time frames that
we expect;
•be unable to satisfy financial obligations to us or others;
•not be able to obtain or maintain all necessary licenses; or
•cease operations.
Any such risks may apply to future products and product candidates we develop, and for which we receive marketing
authorization, such events may result in decreased product sales, which would harm our results of operations and
business.
We rely on third parties as we conduct our clinical trials and some aspects of our research and preclinical studies,
and those third parties may not perform satisfactorily, including failing to meet deadlines for the completion of such
trials, research or testing.
We rely on third parties, such as strategic partners, CROs, clinical data management organizations, medical institutions
and clinical investigators, as we conduct our clinical trials. For example, in China, during 2025 we completed enrollment
for a Phase 3 study of TLX591-Px (marketed as Illuccix in the U.S.) in collaboration with our strategic partner for the
Greater China region, Grand Pharma, and we aim for this study to support future marketing authorization applications for
Illuccix in China. A new drug application was submitted in December 2025 and accepted for review in January 2026 by
the Chinese National Medical Products Administration ("NMPA") Center for Drug Evaluation ("CDE"). We also currently
54
rely and expect to continue to rely on third parties to conduct some aspects of our research and preclinical studies. Any
of these third parties may terminate their engagements with us at any time in accordance with agreements or applicable
laws. If we need to enter into alternative arrangements, our product development activities may be delayed.
Our reliance on these third parties for research and development activities reduces our control over these activities but
does not relieve us of our responsibilities. For example, we remain responsible for ensuring that each of our clinical trials
is conducted in accordance with the general investigational plan and protocols for the trial. Moreover, the FDA requires
us to comply with GCP standards when conducting, recording and reporting the results of clinical trials to ensure that
data and reported results are credible and accurate and that the rights, integrity and confidentiality of trial participants
are protected. The EMA and TGA also require us to comply with comparable standards. Regulatory authorities ensure
compliance with these requirements through periodic inspections of trial sponsors, principal investigators and trial sites.
If we or any of the third parties that we rely on in connection with our clinical trials fail to comply with applicable
requirements, the clinical data generated in our clinical trials may be deemed unreliable and the FDA, EMA or other
comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our
marketing applications. We cannot assure you that upon inspection by a given regulatory authority, such regulatory
authority will determine that any of our clinical trials comply with such requirements. We also are required to register
ongoing clinical trials and post the results of completed clinical trials on a government-sponsored database, such as
ClinicalTrials.gov, within certain timeframes. Failure to do so can result in fines, adverse publicity and civil and criminal
sanctions.
Furthermore, these third parties may also have relationships with other entities, some of which may be our competitors. If
these third parties do not successfully carry out their contractual duties, meet expected deadlines or conduct our clinical
trials in accordance with regulatory requirements or our stated protocols, we will not be able to obtain, or may be
delayed in obtaining, regulatory approvals for our product candidates and will not be able to, or may be delayed in our
efforts to, successfully commercialize our Commercial Products or our product candidates, if approved. In such an event,
our financial results and the commercial prospects for our Commercial Products or product candidates, if approved,
could be harmed, our costs could increase and our ability to generate revenues could be delayed, impaired or foreclosed.
We also expect to rely on other third parties to store and distribute product supplies for our clinical trials. Any
performance failure on the part of such third parties could delay clinical development or regulatory approval of our
product candidates or commercialization of our Commercial Products, producing additional losses and depriving us of
potential product revenue.
In addition, as discussed above, the third parties upon whom we rely to conduct our clinical trials could be negatively
impacted as a result of disruptions caused by, for example, pandemics or epidemics including difficulties in initiating
clinical sites or enrolling participants, travel or quarantine policies, and other factors, including ongoing and future
environmental or geopolitical concerns. If these third parties are so affected, our business prospects and results of
operations could be severely adversely impacted.
We rely on third parties to conduct investigator-sponsored clinical trials of our product candidates. Any failure by a
third-party to meet its obligations with respect to the clinical development of our product candidates may delay or
impair our ability to obtain regulatory approval for our product candidates.
We partly rely on academic and private non-academic institutions to conduct and sponsor clinical trials relating to our
product candidates. We do not control the design or conduct of the investigator-sponsored trials, and it is possible that
the FDA or foreign regulatory authorities will not view these investigator-sponsored trials as providing adequate support
for future clinical trials, whether controlled by us or third parties, for any one or more reasons, including elements of the
design, execution of the trials, safety concerns or other trial results.
Such arrangements will provide us certain information rights with respect to the investigator-sponsored trials, such as
access to and the ability to use and reference the data resulting from the investigator-sponsored trials, including for our
own regulatory submissions and marketing authorization applications. However, we do not have control over the timing
for patient recruitment and reporting of the data from investigator-sponsored trials, nor do we own the data from the
investigator-sponsored trials. If we are unable to confirm or replicate the results from investigator-sponsored trials or if
negative results are obtained, we would likely be further delayed or prevented from advancing clinical development of
our product candidates. Further, if investigators or institutions breach their obligations with respect to the clinical
development of our product candidates, or if the data proves to be inadequate compared to the first-hand knowledge we
might have gained had the investigator-sponsored trials been sponsored and conducted by us, then our ability to rely on
the data from the investigator-sponsored trials in our clinical development plans may be adversely affected.
Additionally, the FDA or foreign regulatory authorities may disagree with the sufficiency of our right to reference the
preclinical, manufacturing or clinical data generated by these investigator-sponsored trials, our right for exclusive
commercial use of the data or our interpretation of preclinical, manufacturing or clinical data from these investigator-
sponsored trials. If so, the FDA or foreign regulatory authorities may require us to obtain and submit additional preclinical,
manufacturing, or clinical data before we may initiate our planned trials and/or may not accept such additional data as
adequate to initiate our planned trials.
We are currently dependent on third parties for the manufacture, distribution and patient dose preparation of our
Commercial Products and product candidates and any difficulties, disruptions, delays or unexpected costs, or the
55
need to find alternative sources, could adversely affect our results of operations, profitability and future business
prospects.
While we have acquired some laboratory capability with Optimal Tracers in Sacramento, IsoTherapeutics in Angleton, and
the facility purchased from ImaginAb, Inc. in Los Angeles, completed Stage 1 of the buildout of our European
manufacturing site in Brussels South, which is operational for selected research and development activities and is good
manufacturing practice ("GMP") accredited for production of Illuccix, progressed the buildout of our TMS North
Melbourne facility for early-stage clinical research and radiopharmaceutical production, and established our first
cyclotron facility in the APAC region in Yokohama, Japan, we currently rely, and expect to continue to rely, on third-party
contract manufacturers to manufacture our Commercial Products and product candidates for our commercial and clinical
use.
Facilities used by our third-party manufacturers may be inspected by the FDA or applicable foreign regulatory authorities
after we submit a marketing application and before potential approval of the product candidate and are also subject to
ongoing periodic unannounced inspections by the FDA or applicable foreign regulatory authorities for compliance with
cGMPs (or similar foreign requirements) and other regulatory requirements following approval. Similar regulations apply
to manufacturers of our Commercial Products and our product candidates for use or sale in foreign countries. We do not
control the manufacturing processes of, and are completely dependent on, our third-party manufacturers for compliance
with the applicable regulatory requirements for the manufacture of our products and product candidates. Third-party
manufacturers may not be able to comply with cGMPs or similar regulatory requirements outside of the U.S.. If our
manufacturers cannot successfully manufacture material that conforms to our specifications and the strict regulatory
requirements of the FDA and any applicable foreign regulatory authority, they will not be able to secure and/or maintain
regulatory approval for their manufacturing facilities. If these facilities are not approved for commercial manufacture or
are not able to maintain approval, we may need to find alternative manufacturing facilities, which could significantly
impact our ability to develop, obtain regulatory approval for or market our Commercial Products or product candidates as
alternative qualified manufacturing facilities may not be available on a timely or cost-efficient basis, or at all. Failure by
any of our manufacturers to comply with applicable cGMPs (and similar foreign requirements) or other regulatory
requirements could result in sanctions being imposed on us or the contract manufacturer, including fines, injunctions,
civil penalties, delays, suspensions or withdrawals of approvals, operating restrictions, interruptions in supply and
criminal prosecutions, any of which could significantly and adversely affect supplies of our Commercial Products or
product candidates and have a material adverse impact on our business, financial condition and results of operations.
We currently have long-term supply agreements with our third-party contract manufacturers to manufacture the clinical
and commercial supplies of our Commercial Products and for our product candidates. Our ability to have our Commercial
Products and product candidates manufactured in sufficient quantities and at acceptable costs to meet our commercial
demand and clinical development needs is dependent on the uninterrupted and efficient operation of our third-party
contract manufacturers’ facilities. Reliance on third-party manufacturers entails risks, including:
•reliance on the third-party for regulatory compliance and quality assurance;
•the possible breach, termination or nonrenewal of a manufacturing agreement by the third-party, including at a time
that is costly or inconvenient to us;
•the possible failure of the third-party to manufacture our Commercial Products or our product candidates according
to our schedule, or at all, including if the third-party manufacturer gives greater priority to the supply of other
products over our Commercial Products or our product candidates, or otherwise does not satisfactorily perform
according to the terms of the manufacturing agreement;
•equipment malfunctions, power outages or other general disruptions experienced by our third-party manufacturers
or distributors to their respective operations and other general problems with a multi-step manufacturing or
distribution process;
•the possible disruptions to supply chain and logistics processes that are required to store, transport, and deliver our
Commercial Products to customers that require timely delivery given the need to inject a dose of our Commercial
Products within a specific window of radioactivity; and
•the possible misappropriation or disclosure by the third-party or others of our proprietary information, including our
trade secrets and know-how.
We currently rely on a single source supplier for our active pharmaceutical ingredient for our Commercial Products and
our related product manufacturing requirements, although additional sources and back-up suppliers are being validated
and implemented. Any performance failure on the part of our existing or future manufacturers could delay clinical
development, regulatory approval or commercialization of our product candidates, if approved. If our suppliers or
contract manufacturers are so affected, our supply chain could be disrupted, our product shipments could be delayed,
our costs could be increased and our business could be adversely affected. If our current contract manufacturers cannot
perform as agreed, we may be required to replace those manufacturers. Although we believe that there are several
potential alternative manufacturers who could manufacture our Commercial Products or our product candidates, we
could incur added costs and delays in identifying and qualifying any such replacement. Consequently, we may not be
able to reach agreement with third-party manufacturers on satisfactory terms, which could negatively impact revenues
56
from sales of our Commercial Products or delay commercialization of any product candidates that are subsequently
approved.
If, because of the factors discussed above, we are unable to have our Commercial Products or our product candidates
manufactured on a timely or sufficient basis, we may not be able to meet clinical development needs or commercial
demand for our Commercial Products or our product candidates or we may not be able to manufacture our Commercial
Products or our product candidates in a cost-effective manner. As a result, we may lose sales, fail to generate projected
revenues or suffer development or regulatory setbacks, any of which could have an adverse impact on our profitability
and future business prospects.
We are currently party to and may seek to enter into additional collaborations, licenses and other similar
arrangements and may not be successful in maintaining existing arrangements or entering into new ones, and even if
we are, we may not realize the benefits of such relationships.
We are currently parties to license and collaboration agreements with a number of pharmaceutical companies and
universities and expect to enter into additional agreements as part of our business strategy. The success of our current
and any future collaboration arrangements will depend heavily on the efforts and activities of our collaborators.
Collaborations are subject to numerous risks, which may include risks that:
•we may not be able to enter into critical strategic collaborations or enter into them on favorable terms;
•collaborators may have significant discretion in determining the efforts and resources that they will apply to
collaborations, and they may not perform their obligations as agreed, expected, or in compliance with applicable
legal requirements;
•collaborators may not pursue development and commercialization of our product candidates or may elect not to
continue or renew development or commercialization programs based on clinical trial results, changes in their
strategic focus due to their acquisition of competitive products or their internal development of competitive
products, availability of funding or other external factors, such as a business combination that diverts resources or
creates competing priorities;
•collaborators may delay clinical trials, provide insufficient funding for a clinical trial program, stop a clinical trial,
abandon a product candidate, repeat or conduct new clinical trials or require a new formulation of a product
candidate for clinical testing;
•collaborators could independently develop, or develop with third parties, products that compete directly or
indirectly with our Commercial Products or product candidates if the collaborators believe that competitive
products are more likely to be successfully developed or can be commercialized under terms that are more
economically attractive than our Commercial Products or product candidates;
•a collaborator with marketing, manufacturing and distribution rights to one or more products may not commit
sufficient resources to or otherwise not perform satisfactorily in carrying out these activities;
•we could grant exclusive rights to our collaborators that would prevent us from collaborating with others;
•collaborators may not properly maintain or defend our intellectual property rights or may use our intellectual
property or proprietary information in a way that gives rise to actual or threatened litigation that could jeopardize or
invalidate our intellectual property or proprietary information or expose us to potential liability;
•disputes may arise between us and a collaborator that cause the delay or termination of the research, development
or commercialization of our current or future product candidates or that results in costly litigation or arbitration that
diverts management attention and resources;
•collaborations may be terminated, which may result in a need for additional capital to pursue further development or
commercialization of the applicable current or future product candidates;
•collaborators may own or co-own intellectual property covering products that result from our collaboration with
them, and in such cases, we would not have the exclusive right to develop or commercialize such intellectual
property;
•disputes may arise with respect to the ownership of any intellectual property developed pursuant to our
collaborations; and
•a collaborator’s sales and marketing activities or other operations may not be in compliance with applicable laws
resulting in civil or criminal proceedings.
Additionally, we may seek to enter into additional collaborations, joint ventures, licenses and other similar arrangements
for the development or commercialization of our product candidates, if approved, due to capital costs required to
develop or commercialize the product candidate, if approved, or manufacturing constraints. We may not be successful in
our efforts to establish such collaborations for our product candidates because our R&D pipeline may be insufficient, our
57
product candidates may be deemed to be at too early of a stage of development for collaborative effort or third parties
may not view our product candidates as having the requisite potential to demonstrate safety and efficacy or significant
commercial opportunity. In addition, we face significant competition in seeking appropriate strategic partners, and the
negotiation process can be time consuming and complex. Further, any future collaboration agreements may restrict us
from entering into additional agreements with potential collaborators. We cannot be certain that, following a strategic
transaction or license, we will achieve an economic benefit that justifies such transaction.
Risks Related to Our Intellectual Property
If we are unable to obtain and/or maintain commercially valuable regulatory exclusivity and intellectual property or to
protect our patents, trademarks, know-how and trade secrets, our ability to successfully commercialize our
Commercial Products and product candidates, if approved, would be adversely impacted.
We rely on effective exclusivity and IP protection and our success will depend in part on our ability to obtain and/or
maintain commercially valuable regulatory exclusivity and patent claims and to protect our patents, trademarks, know-
how and trade secrets. We and our collaboration partners face numerous risks and uncertainties with respect to our
licensed patents and those that may subsequently be licensed or issued to us, including that:
•lodged regulatory filings may not result in intended market or data exclusivity;
•governments may change data and market exclusivity provisions;
•know-how and trade secrets may be published, derived independently by third parties, or otherwise publicly
disclosed, resulting in a loss of protections;
•patent or trademark applications may not result in issued patents or registered trademarks or may take longer than
expected to be issued or registered;
•the claims of any patents or trademarks that are issued or registered may not provide meaningful protection;
•patent term extensions may not be granted or, if granted, may be subject to revision;
•we and our research partners may not be able to develop additional proprietary technologies that are patentable or
otherwise protectable under regulatory exclusivity principles;
•patents issued to us, or our industry partners, may not provide a competitive advantage;
•other companies may challenge our issued patents or trademarks;
•other companies may independently develop similar or alternative technologies to ours or duplicate or design
around our technology;
•other companies may hold patents or trademarks that are relevant to our technology or activities and enforce their
rights against us; and
•if patents are not issued, then the value of our underlying IP rights may be significantly diminished.
Additionally, any information contained in our licensed patents could become part of the public domain, so that it will not
be protected as confidential information or trade secrets. As legal regulations and standards relating to the validity and
scope of regulatory exclusivity and IP continue to evolve around the world, the degree of future protection for our
proprietary rights is uncertain. We may also be subject to arbitrary compulsory licenses or governmental acts reducing IP
protection outside our reasonable control. We may incur significant costs in asserting any patent or trademark IP rights
and in defending legal action against us relating to rights. Such disputes could delay our product development or
commercialization activities. Parties making claims against us may be able to obtain injunctive or other equitable relief
that could prevent us from further developing discoveries or commercializing products or require the payment of
damages or royalties.
In addition, in the event a successful claim of infringement is made against us, we may be required to pay damages and
obtain one or more licenses from the prevailing third-party. If we are not able to obtain these licenses at a reasonable
cost, if it all, we may encounter delays and lose substantial resources while seeking to develop or commercialize
alternative products.
There is a risk that third parties may have IP that is relevant to our proposed activities which could prevent us from
conducting these activities or may require us to license in the third-party’s IP, find alternatives to the third-party IP, or
seek to challenge the third-party IP, either at an administrative stage or through the courts. We may need to acquire or
license IP from third parties to develop and commercialize our own pipeline of IP and products. There is no guarantee
such acquisitions or licenses can be obtained or, if obtained, that they will be on reasonable commercial terms.
Additionally, although we enter into non-disclosure and confidentiality agreements with parties who have access to
patentable aspects of our research and development output, such as our employees, corporate collaborators, outside
58
scientific collaborators, contract research organizations, contract manufacturers, consultants, advisors and other third
parties, there can also be no assurance that any of these parties will not breach confidentiality, or infringe or
misappropriate our IP, which could cause material loss to us.
If we are unable to obtain and maintain patent protection for our products or product candidates and other
discoveries, or if the scope of the patent protection obtained is not sufficiently broad, our competitors could develop
and commercialize products and other discoveries similar or identical to ours, and our ability to successfully
commercialize our products or product candidates and other discoveries may be adversely affected.
Our success depends in large part on our ability to obtain and maintain patent protection in the U.S. and other countries
with respect to our proprietary products and product candidates and other discoveries. We seek to protect our
proprietary position by filing patent applications in the U.S. and abroad related to our products and product candidates
and other discoveries that are important to our business. For a description of our patent portfolio, see “Item 4.
Information on the Company — B. Business Overview.” We intend to continue to apply for patents with claims covering
our key products, product candidates or other discoveries when and where we deem it appropriate to do so.
The patent prosecution process is expensive and time-consuming, and we may not be able to file and prosecute all
necessary or desirable patent applications at a reasonable cost or in a timely manner. It is also possible that we will fail to
identify patentable aspects of our research and development output before it is too late to obtain patent protection. As
such, our intellectual property rights in some countries outside the U.S. can be less extensive than those in the U.S. and
we may not be able to prevent third parties from practicing our inventions in all countries outside the U.S., or from selling
or importing products made using our inventions in and into the U.S. or other jurisdictions. The legal systems of certain
countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual
property protection, particularly those relating to pharmaceuticals or biologics, which could make it difficult for us to stop
the infringement of our patents or marketing of competing products in violation of our proprietary rights generally, which
could result in substantial costs and divert our efforts and attention from other aspects of our business.
In addition, geo-political actions in the U.S. and in foreign countries could increase the uncertainties and costs
surrounding the prosecution or maintenance of our patent applications or those of any current or future licensors and the
maintenance, enforcement or defense of our issued patents or those of any current or future licensors. For example, due
to the Russia-Ukraine conflict, the U.S. and other foreign governments have implemented various economic sanctions
and trade and activity restrictions involving Russia and Belarus. It is possible that additional sanctions and restrictions will
be imposed by the U. S. or other jurisdictions as the Russia-Ukraine conflict continues, and such actions may include
limiting or preventing filing, prosecution, and/or maintenance of patent applications in Russia. Government actions may
also prevent maintenance of issued patents in Russia. These actions could result in abandonment or lapse of our patents
or patent applications, resulting in partial or complete loss of patent rights in Russia. If such an event were to occur, it
could have a material adverse effect on our business.
The patent position of biotechnology and pharmaceutical companies generally is highly uncertain, involves complex legal
and factual questions and has in recent years been the subject of much litigation. As a result, the issuance, scope,
validity, enforceability and commercial value of our patent rights are highly uncertain. Our pending and future patent
applications may not result in patents being issued that protect our product candidates or other discoveries, or which
effectively prevent others from commercializing competitive products and discoveries. Changes in either the patent laws
or interpretation of the patent laws in the U.S. and other countries may diminish the value of our patents or narrow the
scope of our patent protection. The patent positions of companies in the development and commercialization of
pharmaceuticals are particularly uncertain.
The U.S. Supreme Court has ruled on several patent cases in recent years, either narrowing the scope of patent
protection available in certain circumstances or weakening the rights of patent owners in certain situations. Depending
on future actions by the U.S. Congress, the U.S. courts, the U.S. Patent and Trademark Office ("USPTO") and the relevant
law-making bodies in other countries, the laws and regulations governing patents could change in unpredictable ways
that would weaken our ability to obtain new patents or to enforce our existing patents and patents that we might obtain
in the future.
Composition of matter patents for biological and pharmaceutical products and product candidates often provide a strong
form of intellectual property protection for those types of products, as such patents provide protection without regard to
any method of use. We cannot be certain that the claims in our pending patent applications covering compositions of
matter of our product candidates will be considered patentable by the USPTO or by patent offices in foreign countries, or
that the claims in any of our issued patents will be considered valid and enforceable by courts in the U.S. or foreign
countries. Method of use patents protect the use of a product for the specified method. This type of patent does not
prevent a competitor from making and marketing a product that is identical to our product for an indication that is outside
the scope of the patented method. Moreover, even if competitors do not actively promote their product for our targeted
indications, physicians may prescribe these products “off-label.” Although off-label prescriptions may infringe or
contribute to the infringement of method of use patents, the practice is common and such infringement is difficult to
prevent or prosecute.
Publications of discoveries in the scientific literature often lag behind the actual discoveries, and patent applications in
the U.S. and other jurisdictions are typically not published until 18 months after filing, or in some cases not at all.
Therefore, we cannot be certain that we were the first to make the inventions claimed in our patents or pending patent
applications, or that we were the first to file for patent protection of such inventions.
59
Assuming the other requirements for patentability are met, prior to March 2013, in the U.S., the first to invent the claimed
invention was entitled to the patent, while outside of the U.S., the first to file a patent application is entitled to the patent.
In March 2013, the U.S. transitioned to a first-inventor-to-file system in which, assuming the other requirements for
patentability are met, the first inventor to file a patent application is entitled to the patent.
We may be subject to a third-party pre-issuance submission of prior art to the USPTO or become involved in opposition,
derivation, revocation, reexamination, or post-grant or inter partes review or interference proceedings challenging our
patent rights or the patent rights of others. An adverse determination in any such submission, proceeding or litigation
could reduce the scope of, or invalidate, our patent rights, allow third parties to commercialize our discoveries or
products and compete directly with us, without payment to us, or result in our inability to manufacture or commercialize
products without infringing third-party patent rights. In addition, given the amount of time required for the development,
testing and regulatory review of new product candidates, patents protecting such candidates might expire before or
shortly after such candidates are commercialized. Any failure to obtain or maintain patent protection with respect to our
product candidates could have a material adverse effect on our business, financial condition, results of operations and
prospects.
Even if our patent applications issue as patents, they may not issue in a form that will provide us with any meaningful
protection, prevent competitors from competing with us or otherwise provide us with any competitive advantage. Our
competitors may be able to circumvent our patents by developing similar or alternative discoveries or products in a non-
infringing manner.
The issuance of a patent is not conclusive of its inventorship, its scope, validity or its enforceability, and our patents may
be challenged in the courts or patent offices in the U.S. and abroad. Such challenges may result in patent claims being
narrowed, invalidated or held unenforceable, which could limit our ability to stop others from using or commercializing
similar or identical discoveries and products, or limit the duration of the patent protection of our products, product
candidates and discoveries. Given the amount of time required for the development, testing and regulatory review of
new product candidates, patents protecting such candidates might expire before or shortly after such candidates are
commercialized. As a result, our patent portfolio may not provide us with sufficient rights to exclude others from
commercializing products similar or identical to ours.
Our efforts to enforce intellectual property rights around the world may be inadequate to obtain a significant commercial
advantage from the intellectual property that we own or license. Many companies have encountered significant problems
in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries,
particularly certain developing countries, do not favor the enforcement of patents, trade secrets and other intellectual
property protection, particularly those relating to biotechnology or pharmaceutical products, which could make it difficult
for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights
generally. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial costs and divert our
efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted
narrowly, and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate
and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to
enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage
from the intellectual property that we develop or license.
Europe’s Unified Patent Court may in particular present uncertainties for our ability to protect and enforce our patent
rights against competitors in Europe. On June 1, 2023, the EU unitary patent system was launched, providing a single
pan-European Unitary Patent and a new European Unified Patent Court ("UPC"), for litigation involving European patents.
Under the UPC, all European patents, including those issued prior to ratification of the European Patent Package, will by
default automatically fall under the jurisdiction of the UPC. The UPC will provide our competitors with a new forum to
centrally revoke our European patents that have not been opted out of the UPC, and allow for the possibility of a
competitor to obtain pan-European injunctions. It will be several years before we will understand the scope of patent
rights that will be recognized and the strength of patent remedies that will be provided by the UPC. Under the EU unitary
patent system, we will have the right to opt our patents out of the UPC over the first seven years of the court’s existence,
but doing so may preclude us from realizing the benefits of the new unified court.
We may not identify relevant third-party patents or may incorrectly interpret the relevance, scope or expiration of a
third-party patent, which might adversely affect our ability to develop and market our product candidates.
We cannot be certain that we are aware of all third-party patents and pending applications in the U.S. and abroad that
are relevant to or necessary for the commercialization of our product candidates in any jurisdiction. We may not be able
to conduct complete and thorough searches, we may not be able to identify all relevant third-party patents, and we may
not be able to fully predict the scope of the patent claims or the expiration of relevant third-party patent applications that
may issue as patents. The scope of a patent claim is determined by an interpretation of the law, the written disclosure in
a patent and the patent’s prosecution history. Our interpretation of the relevance or the scope of a patent or a pending
application may be incorrect, which may negatively impact our ability to market our product candidates. We may
incorrectly determine that our product candidates are not covered by a third-party patent or may incorrectly predict
whether a third-party’s pending application will issue with claims of relevant scope. Our determination of the expiration
date of any patent in the U.S. or abroad that we consider relevant may be incorrect, which may negatively impact our
ability to develop and market our product candidates. Our failure to identify and correctly interpret relevant patents may
negatively impact our ability to develop and market our product candidates.
60
In addition, the agreements under which we license intellectual property or technology to or from third parties are
complex, and certain provisions in such agreements may be susceptible to multiple interpretations. The resolution of any
contract interpretation disagreement that may arise could narrow what we believe to be the scope of our rights to the
relevant intellectual property or technology or increase what we believe to be our financial or other obligations under the
relevant agreement, either of which could have a material adverse effect on our business, financial condition, results of
operations and prospects. Moreover, if disputes over intellectual property that we have licensed prevent or impair our
ability to maintain our current licensing arrangements on commercially acceptable terms, we may be unable to
successfully develop and commercialize the affected product candidates. Our business also would suffer if any current
or future licensors fail to abide by the terms of the license, if the licensors fail to enforce licensed patents against
infringing third parties, if the licensed patents or other rights are found to be invalid or unenforceable, or if we are unable
to enter into necessary licenses on acceptable terms. Moreover, our licensors may own or control intellectual property
that has not been licensed to us and, as a result, we may be subject to claims, regardless of their merit, that we are
infringing or otherwise violating the licensor’s rights.
Our rights to develop and commercialize our products and product candidates are subject in part to the terms and
conditions of licenses granted to us by others, and the patent protection, prosecution and enforcement for some of
our products and product candidates may be dependent on our licensors.
We currently are reliant upon licenses of certain intellectual property rights and proprietary technologies from third
parties that are important or necessary to the development of our proprietary technologies, including technologies
related to Illuccix and our product candidates. These licenses, and other licenses we may enter into in the future, may
not provide adequate rights to use such intellectual property and proprietary technologies in all relevant fields of use or
in all territories in which we may wish to develop or commercialize technology and product candidates in the future.
Licenses to additional third-party proprietary technology or intellectual property rights that may be required for our
development programs may not be available in the future or may not be available on commercially reasonable terms. In
that event, we may be required to expend significant time and resources to redesign our proprietary technology or
product candidates or to develop or license replacement technology, which may not be feasible on a technical or
commercial basis. If we are unable to do so, we may not be able to develop and commercialize technology and product
candidates in fields of use and territories for which we are not granted rights pursuant to such licenses, which could
harm our competitive position, business, financial condition, results of operations and prospects significantly.
The licensing or acquisition of third-party intellectual property rights is a competitive area, and several more established
companies may pursue strategies to license or acquire third-party intellectual property rights we may consider attractive
or necessary. These established companies may have a competitive advantage over us due to their size, capital
resources and greater clinical development and commercialization capabilities. In addition, companies that perceive us to
be a competitor may be unwilling to assign or license rights to us.
Moreover, some of our owned and in-licensed patents or patent applications or future patents are or may be co-owned
with third parties. If we are unable to obtain an exclusive license to any such third-party co-owners’ interest in such
patents or patent applications, such co-owners may be able to license their rights to other third parties, including our
competitors, and our competitors could market competing products and technology. In addition, we may need the
cooperation of any such co-owners of our patents in order to enforce such patents against third parties, and such
cooperation may not be provided to us. Furthermore, our owned and in-licensed patents may be subject to a reservation
of rights by one or more third parties. Any of the foregoing could have a material adverse effect on our competitive
position, business, financial conditions, results of operations and prospects.
In some circumstances, we may not have the right to control the preparation, filing and prosecution of patent
applications, or to maintain and enforce the patents, covering technology that we license from third parties. In addition,
some of our agreements with our licensors require us to obtain consent from the licensor before we can enforce patent
rights, and our licensor may withhold such consent or may not provide it on a timely basis. Therefore, we cannot be
certain that our licensors or collaborators will prosecute, maintain, enforce and defend such intellectual property rights in
a manner consistent with the best interests of our business, including by taking reasonable measures to protect the
confidentiality of know-how and trade secrets, or by paying all applicable prosecution and maintenance fees related to
intellectual property registrations for any of our products or product candidates and proprietary technologies. We also
cannot be certain that our licensors have drafted or prosecuted the patents and patent applications licensed to us in
compliance with applicable laws and regulations, which may affect the validity and enforceability of such patents or any
patents that may issue from such applications. This could cause us to lose rights in any applicable intellectual property
that we in-license, and as a result our ability to develop and commercialize products or product candidates may be
adversely affected and we may be unable to prevent competitors from making, using and selling competing products.
In addition, our licensors may own or control intellectual property that has not been licensed to us and, as a result, we
may be subject to claims, regardless of their merit, that we are infringing or otherwise violating the licensor’s rights. In
addition, while we cannot currently determine the amount of the royalty obligations we would be required to pay on sales
of future products, if any, the amounts may be significant. The amount of our future royalty obligations will depend on
the technology and intellectual property we use in product candidates that we successfully develop and commercialize,
if any. Therefore, even if we successfully develop and commercialize product candidates, we may be unable to maintain
profitability. In addition, we may seek to obtain additional licenses from our licensors and, in connection with obtaining
such licenses, we may agree to amend our existing licenses in a manner that may be more favorable to the licensors,
including by agreeing to terms that could enable third parties (potentially including our competitors) to receive licenses
61
to a portion of the intellectual property rights that are subject to our existing licenses. Any of these events could have a
material adverse effect on competitive position, business, financial conditions, results of operations, and prospects.
Our technology licensed from third parties may be subject to retained rights.
Any license we may enter into could provide for the retention by the licensor of certain rights under their agreements
with us, including the right to use the underlying technology for non-commercial academic and research use, to publish
general scientific findings from research related to the technology, and to make customary scientific and scholarly
disclosures of information relating to the technology. It is difficult to monitor whether any future licensors will limit their
use of the technology to these uses, and we may incur substantial expenses to enforce our rights to our licensed
technology in the event of misuse.
In addition, the U.S. federal government retains certain rights in inventions produced with its financial assistance under
the Patent and Trademark Law Amendments Act ("the Bayh-Dole Act"). The federal government retains a “nonexclusive,
nontransferable, irrevocable, paid-up license” for its own benefit. The Bayh-Dole Act also provides federal agencies with
“march-in rights.” March-in rights allow the government, in specified circumstances, to require the contractor or
successors in title to the patent to grant a “nonexclusive, partially exclusive, or exclusive license” to a “responsible
applicant or applicants.” If the patent owner refuses to do so, the government may grant the license itself. The Bayh-Dole
Act also imposes other obligations, including the requirement that products covered by the government funded patents
be manufactured in the U.S.. We sometimes collaborate with academic institutions to accelerate our preclinical research
or development. In the future, we may own or license technology which is critical to our business that is developed in
whole or in part with federal funds subject to the Bayh-Dole Act. If the federal government exercises its rights under the
Bayh-Dole Act, our ability to enforce or otherwise exploit patents covering such technology may be adversely affected.
If we fail to comply with our obligations in the agreements under which we license intellectual property rights from
third parties or these agreements are terminated or we otherwise experience disruptions to our business
relationships with our licensors, we could lose intellectual property rights that are important to our business.
We are party to various agreements that we depend on to develop Illuccix, Gozellix and our product candidates and
various proprietary technologies, and our rights to use currently licensed intellectual property, or intellectual property to
be licensed in the future, are or will be subject to the continuation of and our compliance with the terms of these
agreements. For example, under certain of our license agreements we are required to use commercially reasonable
efforts to develop and commercialize product candidates covered by the licensed intellectual property rights, maintain
the licensed intellectual property rights, and achieve certain development milestones, each of which could result in
termination in the event we fail to comply.
In spite of our efforts, our licensors might conclude that we have materially breached our obligations under such license
agreements and might therefore terminate the license agreements, thereby removing or limiting our ability to develop
and commercialize products and technology covered by these license agreements.
Moreover, disputes may arise regarding intellectual property subject to a licensing agreement, including:
•the scope of rights granted under the license agreement and other interpretation-related issues;
•our financial or other obligations under the licensing agreement;
•the extent to which our product candidates, technology and processes infringe on intellectual property of the
licensor that is not subject to the licensing agreement;
•the sublicensing of patent and other rights under our collaboration agreements;
•our rights to transfer or assign the license;
•our diligence obligations under the license agreement and what activities satisfy those diligence obligations;
•the inventorship and ownership of inventions and know-how resulting from the joint creation or use of intellectual
property by our licensors and us and our partners; and
•the priority of invention of patented technology.
In addition, certain provisions in our and our license agreements may be susceptible to multiple interpretations. The
resolution of any contract interpretation disagreement that may arise could narrow what we believe to be the scope of
our rights to the relevant intellectual property or technology, or increase what we believe to be our financial or other
obligations under the agreement, either of which could have a material adverse effect on our business, financial
condition, results of operations and prospects. Moreover, if disputes over intellectual property that we have licensed
prevent or impair our ability to maintain our current licensing arrangements on commercially acceptable terms, we may
be unable to successfully develop and commercialize the affected products or product candidates, which could have a
material adverse effect on our competitive position, business, financial conditions, results of operations and prospects.
We are generally also subject to all of the same risks with respect to protection of intellectual property that we may
62
license as we are for intellectual property that we own, which are described herein. If we or any of our current or future
licensors fail to adequately protect this intellectual property, our ability to commercialize product candidates could suffer.
Issued patents covering our products and product candidates could be found invalid or unenforceable if challenged
in courts or patent offices.
If we or one of our licensing partners initiated legal proceedings against a third-party to enforce a patent covering one or
more of our products or product candidates, the defendant could counterclaim that the asserted patent covering the
relevant product or product candidate is invalid and/or unenforceable. In patent litigation in the U.S., defendant
counterclaims alleging invalidity and/or unenforceability are commonplace. Grounds for a validity challenge could be an
alleged failure to meet any of several statutory requirements, including subject matter eligibility, novelty, obviousness,
written description or enablement. Grounds for an unenforceability assertion could be an allegation that someone
connected with prosecution of the patent withheld material information from the USPTO, or made a misleading
statement, during prosecution with the intent to deceive the USPTO. Third parties may also raise similar claims before
administrative bodies in the U.S. or abroad, even outside the context of litigation. Such mechanisms include re-
examination inter partes review, post grant review, and equivalent proceedings in foreign jurisdictions (e.g., opposition
proceedings). Such proceedings could result in revocation or amendment to our patents in such a way that they no
longer cover our products or product candidates. The outcome following legal assertions of invalidity and
unenforceability is unpredictable. In any patent infringement proceeding, there is a risk that a court will decide that a
patent of ours is invalid or unenforceable, in whole or in part, and that we do not have the right to stop the other party
from using the invention at issue. With respect to the validity question, for example, we cannot be certain that there is no
invalidating prior art, of which we and the patent examiner were unaware during prosecution. There is also a risk that,
even if the validity of such patents is upheld, the court will construe the patent’s claims narrowly or decide that we do not
have the right to stop the other party from using the invention at issue on the grounds that our patent claims do not
cover the invention, or decide that the other party’s use of our patented technology falls under the safe harbor to patent
infringement under 35 U.S.C. §271(e)(1). If a defendant were to prevail on a legal assertion of invalidity and/or
unenforceability, we would lose at least part, and perhaps all, of the patent protection on our products or product
candidates. Such a loss of patent protection would have a material adverse impact on our business. Any of these
occurrences could adversely affect our competitive business position, business prospects and financial condition.
Similarly, if we assert trademark infringement claims, a court may determine that the marks we have asserted are invalid
or unenforceable, or that the party against whom we have asserted trademark infringement has superior rights to the
marks in question. In this case, we could ultimately be forced to cease use of such trademarks.
We may become involved in lawsuits to protect or enforce our patents and other intellectual property rights, which
could be expensive, time-consuming and unsuccessful.
Competitors or commercial supply companies or others may infringe our patents and other intellectual property rights. To
counter infringement, we may be required to file infringement actions, which can be expensive and time-consuming. In
an infringement proceeding, a defendant may assert and a court may agree with a defendant that a patent or other
intellectual property right of ours is invalid or unenforceable (or both), or the factfinder may refuse to stop the other party
from using the allegedly infringed intellectual property at issue.
Even if we establish infringement, the court may decide not to grant an injunction against further infringing activity and
instead award only monetary damages, which may or may not be an adequate remedy. An adverse result in any litigation
could put one or more of our patents or registered trademarks at risk of being invalidated or interpreted narrowly and
could limit our ability to assert our patents or trademark rights against those parties or other competitors and may curtail
or preclude our ability to exclude third parties from making and selling similar or competitive products. Furthermore,
because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk
that some of our confidential information could be compromised by disclosure during this type of litigation.
Third parties may initiate legal proceedings alleging that we are infringing their intellectual property rights, the
outcome of which would be uncertain and could have a material adverse effect on the success of our business.
Our commercial success depends upon our ability and the ability of any current and future collaborators to develop,
manufacture, market and sell our Commercial Products and our product candidates, if approved, and use our proprietary
technologies without infringing the proprietary rights of third parties. We may become party to, or threatened with, future
adversarial proceedings or litigation regarding intellectual property rights with respect to our products or product
candidates and technology, including post-grant proceeding or interference proceedings (patents) or opposition or
cancellation proceedings (trademarks) before the USPTO. Third parties may assert infringement claims against us based
on existing patents or patents that may be granted in the future. No litigation asserting such infringement claims is
currently pending against us, and we have not been found by a court of competent jurisdiction to have infringed a third-
party’s intellectual property rights.
There is a substantial amount of intellectual property litigation in the biotechnology and pharmaceutical industries, and
we may become party to, or threatened with, litigation or other adversarial proceedings regarding intellectual property
rights with respect to our product candidates and Commercial Products. Our product candidates and other proprietary
technologies that we may develop may infringe existing or future patents owned by third parties. Third parties may
assert infringement claims against us based on existing or future intellectual property rights. We may not be aware of
patents that have already been issued and that a third-party, for example, a competitor in the fields in which we are
developing our product candidates, might assert are infringed by our current or future product candidates, including
63
claims to compositions, formulations, methods of manufacture or methods of use or treatment that cover our product
candidates. The coverage of patents is subject to interpretation by the courts, and the interpretation is not always
uniform. If we were sued for patent infringement, we would need to demonstrate that our product candidates,
Commercial Products or methods either do not infringe the patent claims of the relevant patent or that the patent claims
are invalid or unenforceable, and we may not be able to do this. If such patent claims were to survive an invalidity
challenge, and if they were asserted against us, we could incur substantial costs in the resulting litigation, including
possible payment of treble damages for willful infringement and an injunction requiring us to cease sale of our products.
If we are found to infringe or think there is a risk we may be found to infringe, a third-party’s intellectual property rights,
we could be required or choose to obtain a license from such third-party to continue developing, marketing and selling
our products, product candidates and technology. However, we may not be able to obtain any required license on
commercially reasonable terms, or at all. Even if we were able to obtain a license, it could be non-exclusive, thereby
giving our competitors access to the same intellectual property licensed to us. We could be forced, including by court
order, to cease commercializing the infringing intellectual property or product or to cease using the infringing technology.
In addition, we could be found liable for monetary damages. A finding of infringement could prevent us from
commercializing our products or product candidates or force us to cease some of our business operations, and could
divert the time and attention of our technical personnel and management, cause development delays, and/or require us
to develop non-infringing technology, which may not be possible on a cost-effective basis, any of which could materially
harm our business. In the event of a successful claim of infringement against us, we may have to pay substantial
monetary damages, including treble damages and attorneys’ fees for willful infringement, pay royalties and other fees,
redesign our infringing drug or obtain one or more licenses from third parties, which may be impossible or require
substantial time and monetary expenditure. Claims that we have misappropriated the confidential information or trade
secrets of third parties could have a similar negative impact on our business.
Intellectual property disputes could delay or prevent our compassionate use or magisterial preparation access
programs and commercialization, expose us to injunctions, damages (including potential royalty back payments) and
additional costs, and require additional public disclosures that could adversely affect our business and the market
price of our ordinary shares and ADSs.
Intellectual property disputes including patent invalidity actions against 3rd party patents and patent infringement
actions against competitor products are a routine business activity. In February 2026, Telix filed an invalidity action
against an Australian patent owned by the Purdue Research Foundation. Patent disputes are costly, uncertain, and time-
consuming. Patent disputes can result in injunctions, damages (including enhanced damages in some jurisdictions), or
compulsory licensing that disrupt development or commercialization and divert significant management attention and
resources. These risks are heightened where a counterparty seeks injunctive or other equitable relief that could limit or
halt lawful compassionate use or magisterial preparation or commercial supply while a dispute is pending or permanent.
These matters often take years to finalise. Litigation and claims can be expensive and disruptive regardless of their merit,
and we may not prevail. Even if we prevail, such proceedings often involve extensive discovery, may require disclosure
of confidential information, and entail substantial legal expenses and management attention.
Unfavorable outcomes in validity or infringement patent disputes can limit our ability to exclude competitors or operate
without a license, and required licenses may be unavailable on reasonable terms or delayed. Adverse rulings can also
result in damages, ongoing royalty obligations that diminish program economics, or could result in our being enjoined
from importing, making, using, offering to sell, or selling technologies critical to development of our product candidates
and/or commercialization of our Commercial Products.
Public announcements or interim developments may also negatively affect investor perception and the market price of
our ordinary shares and ADSs. Significant developments may necessitate public communications and additional
disclosure in our public filings or offering documents, increasing legal, accounting, and compliance costs and contributing
to volatility in the trading price of our ADSs. Public announcements or interim developments may also negatively affect
investor perception and the market price of our ordinary shares and ADSs. Significant developments may necessitate
public communications and additional disclosure in our public filings or offering documents, increasing legal, accounting,
and compliance costs and contributing to volatility in the trading price of our ADSs.
We may be subject to claims that our employees have wrongfully used or disclosed alleged trade secrets of their
former employers.
Some of our employees may have been previously employed at universities or other biotechnology or pharmaceutical
companies, including our competitors or potential competitors. Although we try to ensure that our employees do not use
the proprietary information or know-how of others in their work for us, we may be subject to claims that we or these
employees have used or disclosed intellectual property, including trade secrets or other proprietary information, of any
such employee’s former employer. Although we have no knowledge of any such claims being alleged to date, if such
claims were to arise, litigation may be necessary to defend against any such claims. If we fail in defending any such
claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel.
Moreover, any such litigation or the threat thereof may adversely affect our reputation, our ability to form strategic
alliances or sublicense our rights to collaborators, engage with scientific advisors or hire employees or consultants, each
of which would have an adverse effect on our business, results of operations and financial condition. Even if we are
successful in defending against such claims, litigation could result in substantial costs and be a distraction to
management.
64
Intellectual property litigation could cause us to spend substantial resources and distract our personnel from their
normal responsibilities.
Even if resolved in our favor, litigation or other legal proceedings relating to intellectual property claims may cause us to
incur significant expenses and could distract our technical and management personnel from their normal responsibilities.
In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or
developments and if securities analysts or investors perceive these results to be negative, it could have a material
adverse effect on the price of our ordinary shares and ADSs. Such litigation or proceedings could substantially increase
our operating losses and reduce the resources available for development activities or any future sales, marketing or
distribution activities.
Further, we may not have sufficient financial or other resources to adequately conduct such litigation or proceedings,
which typically last for years before they are concluded. Because of the expense and uncertainty of litigation, we may
conclude that even if a third-party is infringing our issued patent or trademark rights, any patents that may be issued as
a result of our pending or future patent applications, pending or future trademark applications or other intellectual
property rights, the risk-adjusted cost of bringing and enforcing such a claim or action may be too high or not in the best
interest of our company or our shareholders, or it may be otherwise impractical or undesirable to enforce our intellectual
property rights against some third parties. Some of our competitors may be able to sustain the costs of such litigation or
proceedings more effectively than we can because of their greater financial resources and more mature and developed
intellectual property portfolios. In such cases, we may decide that the more prudent course of action is to simply monitor
the situation or initiate or seek some other non-litigious action or solution. In addition, the uncertainties associated with
the initiation and continuation of patent litigation or other proceedings could have a material adverse effect on our ability
to compete in the marketplace and could compromise our ability to raise the funds necessary to continue our clinical
trials, continue our internal research programs, in-license needed technology or other product candidates, or enter into
development partnerships that would help us bring our product candidates to market. Even if we ultimately prevail in
such claims, the monetary cost of such litigation and the diversion of the attention of our management and scientific
personnel could outweigh any benefit we receive as a result of the proceedings.
Obtaining and maintaining our patent protection depends on compliance with various procedural, documentary, fee
payment and other requirements imposed by governmental patent agencies, and our patent protection could be
reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees, renewal fees, annuity fees and various other governmental fees on patents and/or
applications will be due to the USPTO and various foreign patent offices at various points over the lifetime of the patents
and/or applications. We have systems in place to remind us to pay these fees, and we rely on our outside counsel to pay
these fees when due. Additionally, the USPTO and various foreign patent offices require compliance with a number of
procedural, documentary, fee payment and other similar provisions during the patent application process. We employ
reputable law firms and other professionals to help us comply with such provisions, and in many cases, an inadvertent
lapse can be cured by payment of a late fee or by other means in accordance with rules applicable to the particular
jurisdiction. However, there are situations in which non-compliance can result in abandonment or lapse of the patent or
patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. If such an event were
to occur, it could have a material adverse effect on our business.
Patent terms may be inadequate to protect our competitive position on our product candidates for an adequate
amount of time.
Patent rights are of limited duration. In the U.S., if all maintenance fees are timely paid, the natural expiration of a patent
is generally 20 years after its first effective filing date. Given the amount of time required for the development, testing
and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly
after such product candidates are commercialized. Even if patents covering our product candidates are obtained, once
the patent life has expired for a product, we may be open to competition from biosimilar or generic products. As a result,
our patent portfolio may not provide us with sufficient rights to exclude others from commercializing product candidates
similar or identical to ours. Upon issuance in the U.S., a patent’s life can be increased based on certain delays caused by
the USPTO, but this increase can be reduced or eliminated based on certain delays caused by the patent applicant
during patent prosecution. A patent term extension based on regulatory delay may be available in the U.S.. However,
only a single patent can be extended for each marketing approval, and any patent can be extended only once, for a
single product. Moreover, the scope of protection during the period of the patent term extension does not extend to the
full scope of the claim, but instead only to the scope of the product as approved. Laws governing analogous patent term
extensions in foreign jurisdictions vary widely, as do laws governing the ability to obtain multiple patents from a single
patent family.
Additionally, we may not receive an extension if we fail to exercise due diligence during the testing phase or regulatory
review process, apply within applicable deadlines, fail to apply prior to expiration of relevant patents or otherwise fail to
satisfy applicable requirements. If we are unable to obtain patent term extension or restoration, or the term of any such
extension is less than we request, the period during which we will have the right to exclusively market our product will be
shortened and our competitors may obtain approval of competing products following our patent expiration and may take
advantage of our investment in development and clinical trials by referencing our clinical and preclinical data to launch
their product earlier than might otherwise be the case, and our revenue could be reduced, possibly materially.
65
If our product candidates or any of our future product candidates obtain regulatory approval, additional competitors
could enter the market with generic or similar versions of such products, which may result in a material decline in
sales of our competing products.
Under the Hatch-Waxman Act, a company may submit an ANDA, seeking approval of a generic version of an approved
innovator product. Under the Hatch-Waxman Act, a company may also submit an NDA under section 505(b)(2) of the
FDCA that references the FDA’s prior approval of the innovator product or preclinical studies and/or clinical trials that
were not conducted by, or for, the sponsor and for which the sponsor has not obtained a right of reference. A 505(b)(2)
NDA product may be for a new or improved version of the original innovator product. The Hatch-Waxman Act also
provides for certain periods of regulatory exclusivity, which preclude FDA approval (or in some circumstances, FDA filing
and review) of an ANDA or 505(b)(2) NDA.
In certain circumstances, third parties may submit an ANDA or NDA under Section 505(b)(2) as early as the so-called
“NCE-1” date that is one year before the expiry of the five-year period of New Chemical Entity exclusivity or more
generally four years after NDA approval. The third parties may rely on certain safety and efficacy data of the innovator’s
product, may not need to conduct clinical trials and can market a competing version of a product after the expiration or
loss of patent exclusivity or the expiration or loss of regulatory exclusivity and often charge significantly lower prices.
Upon the expiration or loss of patent protection or the expiration or loss of regulatory exclusivity for a product, the major
portion of revenues for that product may be dramatically reduced in a very short period of time. If we are not successful
in defending our patents and regulatory exclusivities, we will not derive the expected benefit from them.
In addition to the benefits of regulatory exclusivity, an innovator NDA holder may have patents claiming the active
ingredient, product formulation or an approved use of the drug, which would be listed with the product in the FDA
publication “Approved Drug Products with Therapeutic Equivalence Evaluations,” known as the Orange Book. If there are
patents listed in the Orange Book for the applicable, approved innovator product, a generic or 505(b)(2) sponsor that
seeks to market its product before expiration of the patents must include in their applications what is known as a
“Paragraph IV” certification, challenging the validity or enforceability, or claiming non-infringement, of the listed patent or
patents. Notice of the certification must be given to the patent owner and NDA holder and if, within 45 days of receiving
notice, either the patent owner or NDA holder sues for patent infringement, approval of the ANDA or 505(b)(2) NDA is
stayed for up to 30 months.
Accordingly, if any of our product candidates that are regulated as drugs are approved, competitors could file ANDAs for
generic versions of these products or 505(b)(2) NDAs that reference our products. If there are patents listed for such
drug products in the Orange Book, those ANDAs and 505(b)(2) NDAs would be required to include a certification as to
each listed patent indicating whether the ANDA sponsor does or does not intend to challenge the patent. We cannot
predict which, if any, patents in our current portfolio or patents we may obtain in the future will be eligible for listing in
the Orange Book, how any generic competitor would address such patents, whether we would sue on any such patents
or the outcome of any such suit.
If we do not successfully extend the term of patents covering our product candidates under the Hatch-Waxman Act
and similar foreign legislation, our business may be materially harmed.
Depending upon the timing, duration and conditions of FDA marketing approval, if any, of our products or product
candidates, one or more of our U.S. patents may be eligible for patent term extension under the Hatch-Waxman Act. The
Hatch-Waxman Act permits a patent term extension of up to five years for one patent covering an approved product as
compensation for effective patent term lost during product development and the FDA regulatory review process.
However, we may not receive an extension if we fail to apply within applicable deadlines, fail to apply prior to expiration
of relevant patents or otherwise fail to satisfy applicable requirements. Moreover, the length of the extension could be
less than we request. The total patent term, including the extension period, may not exceed 14 years following FDA
approval. Accordingly, the length of the extension, or the ability to even obtain an extension, depends on many factors.
In the U.S., only a single patent can be extended for each qualifying FDA approval, and any patent can be extended only
once and only for a single product. Laws governing analogous patent term extensions in foreign jurisdictions vary widely,
as do laws governing the ability to obtain multiple patents from a single patent family.
If we are unable to obtain a patent term extension for a product or product candidate or the term of any such extension
is less than we request, the period during which we can enforce our patent rights for that product or product candidate,
if any, in that jurisdiction will be shortened and our competitors may obtain approval to market competing products
sooner. As a result, our revenue could be materially reduced.
If we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be
harmed.
In addition to seeking patents for our products, product candidates and other discoveries, we also rely on trade secrets,
including unpatented know-how, technology and other proprietary information, to maintain our competitive position.
Elements of our product candidates, including processes for their preparation and manufacture, involve proprietary
know-how, information, or technology that is not covered by patents, and thus for these aspects we may consider trade
secrets and know-how to be our primary intellectual property. Any disclosure, either intentional or unintentional, by our
employees, the employees of third parties with whom we share our facilities or third-party consultants and vendors that
we engage to perform research, clinical trials or manufacturing activities, or misappropriation by third parties (such as
66
through a cybersecurity breach) of our trade secrets or proprietary information could enable competitors to duplicate or
surpass our technological achievements, thus eroding our competitive position in our market. Because we expect to rely
on third parties in the development and manufacture of our product candidates, we must, at times, share trade secrets
with them. Our reliance on third parties requires us to share our trade secrets, which increases the possibility that a
competitor will discover them or that our trade secrets will be misappropriated or disclosed.
Trade secrets and know-how can be difficult to protect. We seek to protect these trade secrets, in part, by entering into
non-disclosure and confidentiality agreements with parties who have access to them, such as our employees, outside
scientific collaborators, CROs, contract manufacturers, consultants, advisors and other third parties. We also enter into
confidentiality and invention or patent assignment agreements with our employees and consultants. Despite these
efforts, any of these parties may breach the agreements and disclose our proprietary information, including our trade
secrets, and we may not be able to obtain adequate remedies for such breaches. We also may not have entered into
such agreements with each party that may have or has had access to our trade secrets or proprietary technology and
processes. To the extent that we are unable to timely enter into confidentiality and invention or patent assignment
agreements with our employees and consultants, our ability to protect our business through trade secrets and patents
may be harmed. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive
and time-consuming, and the outcome is unpredictable. In addition, some courts inside and outside of the U.S. are less
willing or unwilling to protect trade secrets. If any of our trade secrets were to be lawfully obtained or independently
developed by a competitor, we would have no right to prevent them from using that technology or information to
compete with us. If any of our trade secrets were to be disclosed to or independently developed by a competitor, our
competitive position would be harmed. To the extent inventions are made by a third-party under an agreement that does
not grant us an assignment of their rights in inventions, we may choose or be required to obtain a license.
Not all of our trademarks are registered. Failure to secure those registrations could adversely affect our business.
In total, as of December 31, 2025, we own 26 registered U.S. trademarks, 14 pending U.S. trademark applications, 179
foreign trademarks registered in jurisdictions such as Australia, Europe, China, Brazil and Japan, and 93 pending foreign
trademark applications applied for in jurisdictions such as Australia, Europe, China, Brazil and Japan.
For a description of our registered and pending trademarks, see “Item 4. Information on the Company — B. Business
Overview.”
If we do not secure registrations for our trademarks, we may encounter more difficulty in enforcing them against third
parties than we otherwise would, which could adversely affect our business. During trademark registration proceedings
in the U.S. and foreign jurisdictions, our trademark applications may receive rejections or may face post-registration
rejections. We are given an opportunity to respond to those rejections, but we may not be able to overcome such
rejections. In addition, in the USPTO and in comparable agencies in many foreign jurisdictions, third parties are given an
opportunity to oppose pending trademark applications and to seek to cancel registered trademarks. Opposition or
cancellation proceedings may be filed against our trademarks, and our trademark applications or registrations may not
survive such proceedings.
In addition, any proprietary name we propose to use with our key product candidates in the U.S. must be approved by
the FDA, regardless of whether we have registered it, or applied to register it, as a trademark. The FDA typically
conducts a review of proposed drug names, including an evaluation of potential for confusion with other drug names. If
the FDA (or an equivalent administrative body in a foreign jurisdiction) objects to any of our proposed proprietary drug
names for any of our product candidates, if approved, we may be required to expend significant additional resources in
an effort to identify a suitable proprietary drug name that would qualify under applicable trademark laws, not infringe the
existing rights of third parties and be acceptable to the FDA. Furthermore, in many countries, owning and maintaining a
trademark registration may not provide an adequate defense against a subsequent infringement claim asserted by the
owner of a senior trademark. At times, competitors or other third parties may adopt trade names or trademarks similar to
ours, thereby impeding our ability to build brand identity and possibly leading to market confusion. In addition, there
could be potential trade name or trademark infringement claims brought by owners of other registered trademarks,
common law trademarks (in territories where such claims are permitted under local laws) or trademarks that incorporate
variations of our registered or unregistered trademarks or trade names. If we assert trademark infringement claims, a
court may determine that the trademarks we have asserted are invalid or unenforceable, or that the party against whom
we have asserted trademark infringement has superior rights to the marks in question. In this case, we could ultimately
be forced to cease use of such trademarks, and may be forced to expend resources to identify, protect, and promote a
new trademark to allow for commercialization of certain products.
We may become subject to claims challenging the inventorship or ownership of our patents and other intellectual
property.
We may be subject to claims that former employees, collaborators or other third parties have an interest in our patents or
other intellectual property as an inventor or co-inventor. The failure to name the proper inventors on a patent application
can result in the patents issuing thereon being unenforceable. Inventorship disputes may arise from conflicting views
regarding the contributions of different individuals named as inventors, the effects of foreign laws where foreign
nationals are involved in the development of the subject matter of the patent, conflicting obligations of third parties
involved in developing our product candidates or as a result of questions regarding co-ownership of potential joint
inventions. Litigation may be necessary to resolve these and other claims challenging inventorship and/or ownership.
Alternatively, or additionally, we may enter into agreements to clarify the scope of our rights in such intellectual property.
67
If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual
property rights, such as exclusive ownership of, or right to use, valuable intellectual property. Such an outcome could
have a material adverse effect on our competitive position, business, financial conditions, results of operations, and
prospects. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a
distraction to management and other employees.
Our licensors may have relied on third-party consultants or collaborators or on funds from third parties, such as the U.S.
government, such that our licensors are not the sole and exclusive owners of the patents we in-licensed. If other third
parties have ownership rights or other rights to our in-licensed patents, they may be able to license such patents to our
competitors, and our competitors could market competing products and technology. This could have a material adverse
effect on our competitive position, business, financial conditions, results of operations, and prospects.
In addition, while it is our policy to require our employees and contractors who may be involved in the conception or
development of intellectual property to execute agreements assigning such intellectual property to us, we may be
unsuccessful in executing such an agreement with each party who, in fact, conceives or develops intellectual property
that we regard as our own. The assignment of intellectual property rights may not be self-executing, or the assignment
agreements may be breached, and we may be forced to bring claims against third parties, or defend claims that they
may bring against us, to determine the ownership of what we regard as our intellectual property. Such claims could have
a material adverse effect on our business, financial condition, results of operations, and prospects.
Our proprietary rights may not adequately protect our technologies and product candidates, and do not necessarily
address all potential threats to our competitive advantage.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property
rights have limitations, and may not adequately protect our business, or permit us to maintain our competitive advantage.
The following examples are illustrative:
•others may be able to make products that are the same as or similar to our product candidates but that are not
covered by the claims of the patents that we own or have exclusively licensed;
•others, including inventors or developers of our or our owned or in-licensed patented technologies who may
become involved with competitors, may independently develop similar technologies that function as alternatives or
replacements for any of our technologies without infringing our intellectual property rights;
•we or our licensors or our other collaboration partners might not have been the first to conceive and reduce to
practice the inventions covered by the patents or patent applications that we own or license or will own or license;
•we or our licensors or our other collaboration partners might not have been the first to file patent applications
covering certain of the patents or patent applications that we or they own or have obtained a license, or will own or
will have obtained a license;
•we or our licensors may fail to meet obligations to the U.S. government with respect to in-licensed patents and
patent applications funded by U.S. government grants, leading to the loss of patent rights;
•it is possible that our pending patent applications will not result in issued patents;
•it is possible that there are prior public disclosures that could invalidate our or our licensors’ patents;
•issued patents that we own or exclusively license may not provide us with any competitive advantage, or may be
held invalid or unenforceable, as a result of legal challenges by our competitors;
•our competitors might conduct R&D activities in countries where we do not have patent rights, or in countries where
R&D safe harbor laws exist, and then use the information learned from such activities to develop competitive
products for sale in our major commercial markets;
•ownership, validity or enforceability of our or our licensors’ patents or patent applications may be challenged by
third parties; and
•the patents of third parties or pending or future applications of third parties, if issued, may have an adverse effect
on our business.
Risks Related to Employee Matters and Managing Growth
Our future success depends on our ability to retain key members of our management team and to attract, retain and
motivate qualified personnel.
We are highly dependent on the management, technical and scientific expertise of principal members of our
management and scientific teams, including Christian Behrenbruch, our Managing Director and Group Chief Executive
Officer. Although we have entered into formal employment agreements with our executive officers, these agreements do
68
not prevent them from terminating their employment with us at any time by providing notice within the notice period
specified in such agreements, subject to certain exceptions. We do not maintain “key person” insurance for any of our
executives or other employees. The loss of the services of any of our key employees could impede the achievement of
our research, development, commercialization and other business objectives.
Recruiting and retaining qualified scientific, clinical, manufacturing and sales and marketing personnel is critical to our
success. We may not be able to attract and retain these personnel on acceptable terms given the competition among
numerous pharmaceutical and biotechnology companies for similar personnel. We also experience competition for the
hiring of scientific and clinical personnel from universities and research institutions. In addition, we rely on consultants
and advisors, including scientific and clinical advisors, to assist us in formulating our research and development and
commercialization strategies. Our consultants and advisors may be employed by employers other than us and may have
commitments under consulting or advisory contracts with other entities that may limit their availability to us.
We expect to continue to expand our development and regulatory capabilities, and as a result, we may encounter
difficulties in managing our growth, which could disrupt our operations.
We have experienced rapid growth since our inception in 2017. We expect continued growth in the number of our
employees and the scope of our operations, particularly to continue our clinical operations, preclinical and IND-enabling
studies or studies approved by comparable foreign authorities and to establish regulatory, quality, and manufacturing
supply chain logistics and facility operations.
To manage our anticipated future growth, we will continue to seek to implement and improve our managerial, operational,
and financial systems, expand our facilities, and continue to recruit and train additional qualified personnel. Due to our
limited financial resources and the complexity in managing a company with such anticipated growth, we may not be able
to effectively manage the expansion of our operations or recruit and train additional qualified personnel. In addition, we
are completing the commissioning of a European manufacturing facility in Brussels South and have limited experience in
managing the manufacturing processes necessary for delivering potent therapeutic radioisotopes. The expansion of our
operations may lead to significant costs and may divert our management and business development resources. Any
inability to manage growth could delay the execution of our business plans or disrupt our operations.
In addition, future growth imposes significant added responsibilities on members of management, including: identifying,
recruiting, integrating, maintaining, and motivating new employees; managing our internal development efforts
effectively, including the clinical and FDA, or comparable foreign regulatory authority, and review process for our
Commercial Products and any product candidates, while complying with our contractual obligations to third parties; and
improving our operational, financial and management controls, reporting systems, and procedures. We currently rely, and
for the foreseeable future will continue to rely, in substantial part on certain independent organizations, advisors, and
consultants to provide certain services, including strategic, financial, business development, and research and
development services, as well as certain aspects of regulatory approval and manufacturing. There can be no assurance
that the services of independent organizations, advisors, and consultants will continue to be available to us on a timely
basis when needed or on reasonable terms, or that we can find qualified replacements. In addition, if we are unable to
effectively manage our outsourced activities or if the quality or accuracy of the services provided by consultants, CROs,
or CMOs is compromised for any reason, our preclinical or clinical trials may be extended, delayed, or terminated, and we
may not be able to obtain and/or maintain regulatory approval of our Commercial Products or any of our product
candidates or otherwise advance our business. We cannot assure you that we will be able to manage our existing
consultants or find other competent outside contractors and consultants on economically reasonable terms, or at all.
If we are not able to effectively expand our organization by hiring new qualified employees and expanding our groups of
consultants and contractors, we may experience delays or may not be able to successfully implement the tasks
necessary to further develop and commercialize our Commercial Products and any product candidates we develop and,
accordingly, we may not achieve our research, development, and commercialization goals.
Our business and operations may be materially adversely affected in the event of information technology system
failures or security breaches, and the costs and consequences of implementing data protection measures could be
significant.
Despite the implementation of security measures, our internal computer systems, and those of our CROs and other third
parties on which we rely, are vulnerable to damage from computer viruses, unauthorized access, natural disasters, fire,
terrorism, war and telecommunication and electrical failures. Such systems are also vulnerable to service interruptions or
to security breaches from inadvertent or intentional actions by our employees, third-party vendors and/or business
partners, or from cyber incidents initiated by malicious third parties. Cyber incidents are increasing in their frequency,
sophistication and intensity, and have become increasingly difficult to detect, respond to and recover from. Cyber
incidents could include the deployment of harmful malware, ransomware, denial-of-service attacks, unauthorized access
to or deletion of files, social engineering and other means to affect service reliability and threaten the confidentiality,
integrity and availability of information. Cyber incidents also could include phishing attempts or e-mail fraud to cause
payments or information to be transmitted to an unintended recipient. We could be subject to risks caused by
misappropriation, misuse, leakage, falsification or intentional or accidental release or loss of information maintained in the
information systems and networks of our company, including personal data of our employees, patients and clinical trial
participants. In addition, we face other kinds of risks related to our commercial and personal data, including lost or stolen
devices or other systems (including paper records) that collect and store our personal and commercial information,
including clinical trial data.
69
If such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our
development and commercialization programs and our business operations, whether due to a loss of our trade secrets or
other proprietary information or other similar disruptions. For example, the loss of clinical trial data from completed,
ongoing or planned clinical trials could result in delays in our regulatory approval efforts and significantly increase our
costs to recover or reproduce the data. To the extent that any disruption or security breach were to result in a loss of, or
damage to, our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur
liability, our reputation or competitive position could be damaged, and the further development and commercialization of
our products or product candidates could be delayed or halted. In addition, we may in certain instances be required to
provide notification to individuals or others in connection with the loss of their personal or commercial information.
If a material breach of our security or that of our vendors occurs, our financial or other confidential information could be
compromised and could adversely affect our business or result in legal proceedings. In addition, the cost and operational
consequences of implementing further data protection measures could be significant. The development and
maintenance of these systems, controls and processes is costly and requires ongoing monitoring and updating as
technologies change and efforts to overcome security measures become more sophisticated. Moreover, the possibility of
these events occurring cannot be eliminated entirely.
Our employees, independent contractors, consultants, clinical trial investigators, collaborators and vendors may
engage in misconduct or other improper activities, including non-compliance with regulatory standards and/or
requirements and insider trading, which could cause significant liability for us and harm our reputation.
We are exposed to the risk of fraud or other misconduct by our employees, independent contractors, consultants,
collaborators and vendors. Misconduct by these partners could include intentional, reckless and/or negligent conduct or
unauthorized activities that violate FDA regulations or similar regulations of comparable foreign regulatory authorities;
provide inaccurate information to the FDA or comparable foreign regulatory authorities; fail to comply with manufacturing
standards, federal and state healthcare fraud and abuse laws and regulations and similar laws and regulations
established and enforced by comparable foreign regulatory authorities; fail to comply with state drug pricing
transparency filing requirements; fail to report financial information or data accurately; or fail to disclose unauthorized
activities to us. Employee misconduct could also involve the improper use of information obtained in the course of clinical
trials, which could result in regulatory sanctions and serious harm to our reputation. This could include violations of
HIPAA, other U.S. federal and state laws, and requirements of foreign jurisdictions, including GDPR. We are also exposed
to risks in connection with any insider trading violations by employees or others affiliated with us. It is not always
possible to identify and deter employee or third-party misconduct, and the precautions we take to detect and prevent
these activities may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from
significant penalties, governmental investigations or other actions or lawsuits stemming from a failure to be in
compliance with such laws, standards, regulations, guidance or codes of conduct. If any such actions are instituted
against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a
significant impact on our business and results of operations, including the imposition of significant fines or other
sanctions.
Legal claims and proceedings could adversely impact our business.
We have been in the past the subject of employment-related claims, and may in the future be a party to employment-
related litigation, and any future litigation related to such actions could materially adversely affect us. We consider our
historical experiences with such claims and proceedings to be in the normal course of our business or typical for our
industry; however, it is difficult to assess the outcome of these matters, and we may not prevail in any future
proceedings or litigation. Regardless of their merit, any threatened or actual claims or proceedings can require significant
time and expense to investigate and defend. Since litigation is inherently uncertain, there is no guarantee that we will be
successful in defending ourselves against such claims or proceedings, or that our assessment of the materiality of these
matters, including any reserves taken in connection therewith, will be consistent with the ultimate outcome of such
matters.
Risks Related to Sustainability
We may experience sustainability risks, including: physical climate-related risks such as extreme weather events
that could disrupt internal operations, suppliers, and logistics, and increase operational costs; resource and energy
risks such as increasing energy prices, regulatory actions and increased resource efficiency standards that may
increase operating expenses and capital expenditure for facility upgrades; and, non-compliance with increased
social and governance regulations that could lead to fines, reputational damage, and increased compliance costs.
The Group identifies and manages sustainability risks through a structured process aligned with global reporting
standards and investor expectations. In 2025, the Group completed a Double Materiality Assessment ("DMA"), which
evaluates both impact materiality (how operations affect the environment and society) and financial materiality (how
these impacts influence enterprise value). This assessment informs strategic priorities, and we have identified the
following risk areas:
•Physical Climate Risk: Extreme weather events such as floods, storms, and heatwaves may disrupt suppliers and
logistics, increase operational costs, and threaten facility safety.
70
•Resource and Energy Risk: Rising energy prices and regulatory actions on resource efficiency standards may
increase operating expenses and capital requirements for facility upgrades and sustainable alternatives.
•Social and Governance Risks: Non-compliance with health and safety, end-user data privacy, and anti-corruption
regulations may result in fines, reputational damage, and increased compliance costs.
These risks and others are described in greater detail in the following risk factors and in our Sustainability report.
Physical Climate Risks
Increasing frequency and severity of climate-related events could disrupt our manufacturing, key suppliers, logistics
and workforce and adversely affect our business and we may not be sufficiently resilient to physical climate risks,
requiring significant investment and potentially constraining our site selection.
Our global operations are exposed to acute and chronic physical risks associated with climate change, including more
frequent and severe storms, floods, heatwaves, wildfires, and other extreme weather events. These events can damage
or impair our manufacturing facilities, laboratories, warehouses, clinical trial sites, data centers and office locations, and
can disrupt utilities and critical infrastructure on which we depend. They may also interrupt our logistics and distribution
networks, including air, sea and land transport routes, and compromise the ability of our employees and contractors to
safely travel to and from company and third‑party sites. Any such disruption could delay the manufacture, testing,
release and delivery of our Commercial Products and regulatory trial materials, increase operating and capital costs
surrounding repairs, hardening, redundancy and insurance, and result in product shortages, lost revenue and damage to
our relationships with customers, healthcare providers and partners. If these climate‑related events increase in frequency
or severity or occur in regions that are strategically important to us, our business, financial condition and results of
operations could be materially and adversely affected.
Certain of our existing facilities and those of our critical suppliers and contract manufacturers are located in regions that
are exposed to increased physical climate risks. Enhancing the resilience of these sites through elevation, hardening,
redundancy in utilities, flood protection, enhanced cooling or other measures may require significant capital expenditures
and extended project timelines. In addition, as we consider new manufacturing, research or distribution sites, physical
climate risk is increasingly a factor in site selection. Suitable locations that offer both operational advantages and
acceptable climate risk profiles may be limited, more expensive, or subject to competing demand from other companies.
If we are unable to adequately assess and mitigate climate‑related physical risks at our existing and future sites, we
could experience higher operating and transportation costs, more frequent disruptions, and constraints on our ability to
expand capacity in a timely and cost‑effective manner.
Water scarcity and wastewater management regulations may adversely affect our manufacturing operations, quality
controls and expansion plans.
Many of our Commercial Products and active pharmaceutical ingredients (“APIs”) are manufactured using water‑intensive
processes which require access to reliable sources of water of appropriate quality, as well as adequate wastewater
treatment and discharge infrastructure. In some regions where we operate or may expand, climate change is contributing
to water scarcity, drought, flooding, or changes in water quality and regulatory requirements. Governments in the
countries where we manufacture our products may impose stricter limits on industrial water use, require additional
investments in water treatment, or restrict new permits and expansions in water‑stressed areas. In some regions, tighter
regulation of industrial water use, wastewater discharge, waste management, and renewable energy procurement is
affecting the feasibility and cost of pharmaceutical operations. We may face requirements to invest in additional on‑site
water or waste infrastructure, secure long‑term renewable energy contracts, or demonstrate compliance with local
climate and environmental standards as a condition to permitting or operating. If we are unable to identify targets or sites
that meet our operational needs and ESG expectations, or if environmental and resource constraints materially increase
the cost or complexity to secure sufficient, reliable and compliant water and wastewater capacity on acceptable terms,
we may be forced to reduce production volumes, reconfigure or relocate manufacturing operations, or materially alter
strategic plans or expansions. We may also incur higher costs to treat, recycle or transport water and wastewater, or to
install alternative water technologies. Any of these developments could adversely affect our manufacturing reliability,
product quality, cost structure and growth strategy.
Climate-related disruptions to transportation and cold-chain logistics could impair the distribution and integrity of
our products.
Many of our products, drug substances and intermediates, including temperature‑sensitive biologics and, where
applicable, radiopharmaceuticals and other hazardous materials, must be stored and transported under strict
temperature‑controlled conditions and delivered within defined time windows. Climate‑related impacts and extreme
weather events, including, but not limited to heatwaves, arctic storms, tropical storms, flooding, and potential resulting
infrastructure failure, can disrupt air and ground transportation, reduce availability of cargo capacity, and increase the
risk of temperature excursions or delays during shipping. We substantially rely on third‑party logistics providers, specialty
couriers, and in some cases just‑in‑time manufacturing and delivery models. Any failure in transport infrastructure or
cold‑chain logistics due to physical climate events, power outages, or related issues could result in spoilage, loss, or
delays of finished products or clinical supplies. This could lead to product shortages, recalls, regulatory non‑compliance,
contractual penalties, or damage to our reputation, any of which could materially and adversely affect our business.
71
Resource and Energy Risks
Our operations depend on reliable access to energy and increasing energy costs, supply disruptions and
decarbonization pressures could adversely affect our business.
Our manufacturing, research, information technology systems, and office operations require significant and growing
amounts of energy to operate. Climate change and the global transition to a lower‑carbon economy are contributing to
energy price volatility, changes in energy mix, and, in some regions, constraints on grid reliability and capacity. We also
anticipate higher energy demand for temperature control in our facilities and across our distribution network as global
average temperatures rise. We are implementing measures to reduce greenhouse gas emissions, which may include
entering into virtual power purchase agreements and power purchase agreements, investing in on‑site renewable energy
installations, and improving energy usage efficiency. These initiatives require capital and may not be available on
acceptable terms in all jurisdictions, particularly where competition for renewable energy is increasing. If we experience
sustained energy price increases, energy supply interruptions, or are unable to execute our energy strategy effectively,
our capital expenditures and operating costs could increase and our ability to meet sustainability commitments could be
impaired, causing our business and results of operations to be adversely affected.
Limited availability or regulatory constraints on critical raw materials, including biologics and other specialized
inputs, could increase our costs and disrupt production.
We rely on a wide range of raw materials, packaging materials and specialized inputs sourced from third parties
worldwide. These include, but are not limited to, materials derived from biological sources (such as components used in
certain quality control tests), as well as materials that are manufactured using fossil fuel‑based feedstocks or complex
chemical processes. Environmental degradation, biodiversity loss, such as declines in certain species used in
pharmaceutical testing or production, regulatory restrictions, and changes in conservation or harvesting policies may
reduce the availability or increase the cost of some of these inputs. If key materials become scarce, become subject to
new environmental or animal‑welfare regulations, or are concentrated among a small number of suppliers able to meet
evolving ESG standards, we may incur higher procurement costs, need to invest in reformulation or alternative
technologies, or experience delays or interruptions in manufacturing. Any significant interruption in the supply of critical
biologics or raw materials could adversely affect our ability to manufacture and deliver products on a timely basis and
could negatively impact our financial performance.
Transitioning our operations and supply chain to a lower‑carbon model may require substantial investment and may
not proceed as planned.
Regulators, investors, customers and other stakeholders increasingly expect pharmaceutical companies to reduce
greenhouse gas emissions, including Scope 3 emissions across their supply and value chains. Meeting these
expectations may require us to modify processes, invest in new equipment or technologies, change product designs or
packaging, restructure logistics networks, and shift to lower‑carbon energy and materials. For certain specialized
technologies, such as energy‑intensive manufacturing steps or generation of medical isotopes where applicable,
economical low‑carbon alternatives may not yet be available or scalable. If we are unable to implement our
decarbonization initiatives on commercially reasonable terms, or if regulation accelerates faster than technology and
markets evolve, we may face higher compliance costs, potential carbon pricing or taxes, and constraints on operations or
expansion in certain markets. Failure to meet our stated climate or energy targets could also adversely affect our
reputation and our relationships with investors, customers and partners.
We generate hazardous and, at times, radioactive and biologically active waste where evolving environmental, health
and safety requirements may increase our costs and liabilities.
Our manufacturing, research and development, and where relevant, radiopharmaceutical operations, generate
hazardous, chemical, biological and, in some cases, radioactive waste that must be collected, handled, stored,
transported, treated and disposed of in compliance with complex and evolving environmental, health and safety laws and
regulations. Regulators may impose more stringent requirements over time, including with respect to waste minimization,
tracking, treatment technology, emissions limits and long‑term disposal obligations. As the scale and complexity of our
operations expand, the volume of such waste we generate is likely to increase. We may need to invest in additional
on‑site infrastructure, contract with specialized third‑party waste management providers, or adapt to changes in
available disposal capacity or technology. Any failure of our ability to properly manage hazardous or radioactive materials
and waste could result in regulatory enforcement, cleanup obligations, operational restrictions, civil or criminal penalties,
or claims for personal injury, property damage, or environmental harm. Such events could materially and adversely affect
our business, financial condition, results of operations and reputation.
Social and Governance Risks
Failure to meet evolving environmental, social and governance disclosure expectations and regulatory requirements
could adversely affect our access to capital, valuation and reputation.
Evolving expectations and regulatory requirements relating to ESG matters, including climate-related issues, could
adversely affect our business, financial condition and prospects. Regulators and standard-setting bodies in the European
Union and other jurisdictions in which we operate are implementing or may implement new requirements regarding
climate-related and broader ESG disclosures, including with respect to greenhouse gas emissions, climate risk
72
governance and supply chain transparency. Investors, lenders, customers and other stakeholders are also increasingly
relying on ESG ratings, benchmarks and voluntary disclosures in making investment, procurement and business
decisions. Collecting, verifying and reporting ESG data across our global operations and value chain is exceptionally
complex and resource-intensive while being subject to evolving standards and methodologies. Failure to adequately
integrate ESG considerations, including climate resilience, resource use, human rights and compliance with
environmental and safety regulations, into our governance structures, risk management processes, strategic planning,
capital allocation and expansions could lead to suboptimal decisions, stranded assets, unanticipated compliance and
remediation costs, and underperforming acquisitions. If our ESG-related disclosures or actions are perceived as
incomplete, inaccurate, inconsistent or not aligned with emerging regulatory, customer or investor expectations, we
could face regulatory scrutiny, investigations, sanctions, fines, litigation, reduced access to capital or less favorable
financing terms, and damage to our reputation, and our ability to attract and retain certain investors could be adversely
affected.
ESG practices, especially regarding diversity, equity and inclusion (“DEI”), have been increasingly subject to political
controversy in the United States in recent years. Our policies and practices regarding DEI and other ESG-related matters,
including those that may be required by non-U.S. law, may expose us to legal, reputational and other risks, including anti-
ESG and anti-DEI-related orders, investigations, legislation, litigation, media coverage and scrutiny, boycotts and
negative publicity from investors and other stakeholders. We cannot predict what regulatory or other changes may occur
in the future as a result of this controversy, and we may not be able to meet any conflicting expectations of some or all of
our investors, customers, employees and other third parties (including governmental entities and officials and non-
governmental organizations) regarding various aspects of our business, including with respect to DEI and other ESG
matters.
Failure to manage ESG risks across our operations, workforce and global value chain could result in legal, operational
and reputational harms.
Inadequate management of ESG risks across our operations, workforce and global value chain, including labor, human
rights, modern slavery and climate-related health and safety risks, could adversely affect our operations, reputation and
competitive position. We operate in, and source products and services from, multiple countries, including regions with
higher risks relating to labor practices, human rights and modern slavery. We substantially rely on third-party suppliers,
contract manufacturers, distributors, logistics providers and other partners, some of which in turn rely on their own
complex supply chains, and we may not have complete visibility into or control over their practices. Stakeholders and
regulators are increasingly scrutinizing companies’ efforts to prevent forced labor, child labor, unsafe working conditions
and other human rights abuses in their operations and supply chains. Our employees and contractors work in
manufacturing plants, laboratories, offices, distribution centers and in the field, and climate change can exacerbate
occupational health and safety risks, including heat stress, vector-borne diseases, air quality issues and extreme weather
events. Inadequate planning for emergency response, workplace adaptation, remote work capabilities or business
continuity in the face of climate-related events could lead to increased absenteeism, reduced productivity, higher
healthcare and insurance costs, and difficulties recruiting and retaining talent, particularly in climate-stressed regions.
Public and private healthcare systems, hospital groups, group purchasing organizations and other customers are
increasingly considering suppliers’ environmental and social performance, including carbon footprint, waste and water
management, supply chain practices and human rights, when making procurement and reimbursement decisions, and
some tenders and contracts include ESG criteria or require detailed ESG disclosures.
If we or our partners fail to meet applicable modern slavery, human rights, labor or climate-related health and safety
standards, or if we are unable to demonstrate ESG performance, climate risk management and supply chain transparency
that meet the expectations of key stakeholders, we could face regulatory investigations, fines, litigation, supply
disruptions, product boycotts, loss of customers, reduced pricing power, exclusion from preferred supplier lists and
restrictions on access to certain markets, and our reputation and demand for our products could be adversely affected.
Risks Related to an Investment in the ADSs
An active and liquid market for our securities may not continue to be developed or sustained, which could harm the
market price of the ADSs.
While our ordinary shares have been listed on the ASX since 2017, prior to the November 2024 listing of the ADSs on
Nasdaq, there was no public market on a U.S. national securities exchange for our ordinary shares or ADSs. Given the
limited trading history, an active trading market for the ADSs may not continue to be developed or sustained. In the
absence of an active trading market for the ADSs, investors may not be able to sell their ADSs.
Future sales of ordinary shares or ADSs by existing holders could depress the market price of the ordinary shares or
ADSs.
Sales of a substantial number of shares or ADSs in the public market, or the perception that such sales could occur,
could adversely affect the market price of our ordinary shares or ADSs. As of December 31, 2025, we had 338,777,049
outstanding ordinary shares, and approximately 31,744,502 in ordinary shares underlying outstanding share options and
other equity securities convertible into or exercisable for ordinary shares. In addition, as of December 31, 2025, there
were approximately 26,230,831 ordinary shares underlying outstanding Convertible Bonds, which may be converted at
73
the option of the holders, subject to the conditions in the trust deed, at any time on or after September 9, 2024, at an
initial conversion price of A$24.78 per ordinary share, subject to adjustment.
Ordinary shares underlying these securities may become eligible for sale in the public market in the future, subject to
certain legal and contractual limitations. Sales of a large number of the ordinary shares in the public market could
depress the market price of the ordinary shares or the ADSs. If these additional ordinary shares are sold, or if it is
perceived that they will be sold, in the public market, the trading price of the ordinary shares and ADSs could decline
substantially, which could impair our ability to raise additional capital through the issuance of ordinary shares, ADSs or
other securities in the future.
Our shareholders may experience dilution if we issue ordinary shares or ADSs in future financings, and, as a result,
the price of the ordinary shares or ADSs may decline.
We may from time-to-time issue additional ordinary shares or ADSs and such issuance may occur at a discount from the
trading price of the ordinary shares or ADSs. Additionally, we have in the past issued debt securities convertible into
equity, and we may do so again in the future. For example, in July 2024, we issued the Convertible Bonds, which may be
converted into ordinary shares. As a result, holders of the ADSs could experience immediate dilution upon the issuance
of any of our ordinary shares, including as a result of the conversion of some or all of the Convertible Bonds. As
opportunities present themselves, we may enter into financing or similar arrangements in the future, including the
issuance of debt securities, preference shares or shares. If we issue ordinary shares or other equity or equity-linked
securities, holders of ADSs would experience additional dilution and, as a result, the trading price of the ordinary shares
or ADSs may decline.
The rights of holders of ADSs to participate in any future preferential subscription rights offering or to elect to
receive dividends in ordinary shares may be limited, which may cause dilution to their holdings.
The deposit agreement provides that the depositary will not make rights available to holders of ADSs unless the
distribution to ADS holders of both the rights and any related securities are either registered under the Securities Act or
exempted from registration under the U.S. Securities Act of 1933, as amended ("Securities Act"). If we offer holders of
our ordinary shares the option to receive dividends in either cash or shares, under the deposit agreement the depositary
may require satisfactory assurances from us that extending the offer to holders of ADSs does not require registration of
any securities under the Securities Act before making the option available to holders of ADSs. We are under no obligation
to file a registration statement with respect to any such rights or securities or to endeavor to cause such a registration
statement to be declared effective. Moreover, we may not be able to establish an exemption from registration under the
Securities Act. Accordingly, ADS holders may be unable to participate in our rights offerings or to elect to receive
dividends in shares and may experience dilution in their holdings. In addition, if the depositary is unable to sell rights that
are not exercised or not distributed or if the sale is not lawful or reasonably practicable, it will allow the rights to lapse, in
which case holders of ADSs will receive no value for these rights.
Our principal shareholders and management own a significant percentage of our ordinary shares and may be able to
exert significant influence over matters subject to shareholder approval.
As of December 31, 2025, our executive officers, directors, holders of 5% or more of our outstanding equity interests and
their respective affiliates beneficially owned approximately 7.71% of our outstanding ordinary shares. These shareholders
may be able to determine all matters requiring shareholder approval and they may have interests that differ from other
shareholders and may be adverse to the interests of other shareholders. For example, these shareholders may be able to
propose elections of directors, amendments of our organizational documents, or approval of any merger, sale of assets,
or other major corporate transaction, and may be able to exert significant influence over matters subject to shareholder
approval. Please see "Item 7. Major Shareholders and Related Party Transactions - A. Major Shareholders" for more
information on the beneficial ownership of our executive officers, directors and holders of 5% or more of our outstanding
equity interests.
ADS holders may not be entitled to a trial by jury with respect to claims arising under the deposit agreement, which
could result in less favorable outcomes to the plaintiffs in any such action.
The deposit agreement governing our ADSs provides that, to the fullest extent permitted by applicable law, ADS holders,
including holders who acquire ADSs in the secondary market, irrevocably waive the right to a trial by jury for any claim
they may have against us or the depositary arising out of or relating to the deposit agreement, the shares or the ADSs,
including claims under U.S. federal securities laws.
If we or the depositary opposed a jury trial demand based on the waiver, the court would determine whether the waiver
was enforceable based on the facts and circumstances of that case in accordance with the applicable state and federal
law. To our knowledge, the enforceability of a contractual pre-dispute jury trial waiver in connection with claims arising
under the federal securities laws has not been finally adjudicated by the U.S. Supreme Court. If this jury trial waiver
provision is prohibited by applicable law, an action could nevertheless proceed under the terms of the deposit agreement
with a trial by jury. However, we believe that a contractual pre-dispute jury trial waiver provision is generally enforceable,
including under the laws of the State of New York, which govern the deposit agreement, by a federal or state court in the
City of New York, which has non-exclusive jurisdiction over matters arising under the deposit agreement. In determining
74
whether to enforce a jury trial waiver provision, New York courts and federal courts will consider whether the visibility of
the jury trial waiver provision within the agreement is sufficiently prominent such that a party has knowingly waived any
right to trial by jury. We believe that this is the case with respect to the deposit agreement and the ADSs. It is advisable
that potential owners or holders of ADSs consult legal counsel regarding the jury waiver provision before acquiring any
ADS(s) and thereby becoming subject to the terms of the deposit agreement.
If any owner or holder of our ADSs, including purchasers of ADSs in secondary market transactions, brings a claim
against us or the depositary in connection with matters arising under the deposit agreement or the ADSs, including
claims under U.S. federal securities laws, such owner or holder may incur increased costs of bringing a claim and may not
be entitled to a trial by jury with respect to such claims, which may have the effect of limiting and discouraging lawsuits
against us or the depositary. If a lawsuit is brought against us or the depositary under the deposit agreement, it may be
heard only by a judge of the applicable trial court, which would be conducted according to different civil procedures and
may result in different outcomes than a trial by jury would have had, including results that could be less favorable to the
plaintiffs in any such action. The deposit agreement governing our ADSs provides that any legal suit, action or
proceeding against or involving us brought by the depositary or any holder or beneficial owner of ADSs, arising out of or
based upon the deposit agreement, the ADSs, the ADRs or the transactions contemplated therein or thereby, may be
instituted only in any state or federal court in New York, New York. This forum provision may increase costs to owners or
holders of ADRs and limit their ability to bring a claim in a judicial forum that they find favorable for disputes with the
depositary or us, or the depositary’s or our respective directors, officers or employees, which may discourage such
lawsuits against the depositary, us and the depositary’s and our respective directors, officers or employees. However, it
is possible that a court could find this choice of forum provision to be inapplicable or unenforceable. The enforceability of
similar choice of forum provisions has been challenged in legal proceedings. Any legal suit, action or proceeding against
or involving the depositary brought by us, arising out of or based upon the deposit agreement, the ADSs, the ADRs or the
transactions contemplated therein or thereby, may only be instituted in a state or federal court in New York, New York.
No condition, stipulation or provision of the deposit agreement or ADSs serves as a waiver by any holder or beneficial
owner of ADSs or by us or the depositary of compliance with any provision of U.S. federal securities laws and the rules
and regulations promulgated thereunder.
Limitations in the deposit agreement may not be effective to waive claims against the Company based on compliance
with the federal securities laws.
Although the deposit agreement provides a waiver of trial by jury as described above, we have been advised that no
condition, stipulation or provision of the deposit agreement or ADSs can serve as a waiver by any owner or holder of
ADSs or by us or the depositary of compliance with any substantive provision of the U.S. federal securities laws and the
rules and regulations promulgated thereunder.
The market price and trading volume of the ADSs may be volatile and may be affected by economic conditions
beyond our control.
The market price of the ADSs may be highly volatile and subject to wide fluctuations. The stock market in general, and
the market for biopharmaceutical companies in particular, has experienced extreme volatility that has often been
unrelated to the operating performance of particular companies. In addition, the trading volume of the ADSs may
fluctuate and cause significant price variations to occur. If the market price of the ADSs declines significantly, you may
be unable to resell the ADSs at or above the purchase price, if at all. We cannot assure you that the market price of the
ADSs will not fluctuate or significantly decline in the future.
Some specific factors that could negatively affect the price of the ADSs or result in fluctuations in their price and trading
volume include:
•adverse results or delays in our preclinical studies or clinical trials;
•reports of adverse events or other negative results in clinical trials of third parties’ product candidates that target
our products’ or product candidates’ target indications;
•an inability for us to obtain additional funding on reasonable terms or at all;
•any delay in submitting an IND, BLA or NDA (or similar foreign application) for our product candidates and any
adverse development or perceived adverse development with respect to the FDA’s (or comparable foreign
regulatory authority’s) review of that IND, BLA or NDA (or similar foreign application);
•failure to develop successfully and commercialize our products and product candidates;
•announcements we make regarding our current products and product candidates, acquisition of potential new
products/product candidates and companies and/or in-licensing;
•failure to maintain our existing license arrangements or enter into new licensing and collaboration agreements;
•failure by us or our licensors to prosecute, maintain or enforce our intellectual property rights;
•changes in laws or regulations applicable to current and future products;
75
•inability to obtain adequate clinical or commercial supply for our product candidates or the inability to do so at
acceptable prices;
•adverse regulatory decisions, including failure to reach agreement with applicable regulatory authorities on the
design or scope of our planned clinical trials;
•failure to obtain and maintain regulatory exclusivity for our products and product candidates;
•regulatory approval or commercialization of new products or other methods of treating our target disease
indications by our competitors;
•failure to meet or exceed financial projections we may provide to the public or to the investment community;
•publication of research reports or comments by securities or industry analysts;
•the perception of the pharmaceutical and biotechnology industries, and especially the radiopharmaceutical industry,
by the public, legislatures, regulators and the investment community;
•announcements of significant acquisitions, strategic partnerships, joint ventures or capital commitments by us, our
strategic collaboration partners or our competitors;
•disputes or other developments relating to proprietary rights, including patents, litigation matters and our ability to
obtain patent protection for our technologies;
•additions to or departures of our key scientific or management personnel;
•significant lawsuits, including patent or shareholder litigation, against us;
•changes in the market valuations of similar companies;
•fluctuations of exchange rates between the U.S. dollar and the Australian dollar;
•changes in trading volume of ADSs on Nasdaq and of our ordinary shares on the ASX;
•sales or perceived potential sales of the ADSs or ordinary shares by us, our directors, executive officers or our
shareholders in the future;
•announcement or expectations of additional financing efforts; and
•conditions in the U.S. or Australian financial markets or changes in general economic conditions.
ADS holders are not our shareholders and do not have shareholder rights.
JPMorgan Chase Bank, N.A., as depositary, issues, registers and delivers the ADSs. After purchasing an ADS, holders
hold ADSs with underlying ordinary shares in an Australian publicly listed company. ADS holders are not treated as our
shareholders and do not have shareholder rights. The depositary is the holder of our ordinary shares underlying the
ADSs. Holders of ADSs have ADS holder rights, which are solely contractual in nature. A deposit agreement among us,
the depositary, ADS holders, and the beneficial owners of ADSs, sets out ADS holder rights as well as the rights and
obligations of the depositary. New York law governs the deposit agreement and the ADSs. We and the depositary may
amend or terminate the deposit agreement without the ADS holders’ consent in a manner that could prejudice ADS
holders. For a description of ADS holder rights, see “Item 12. Description of Securities Other than Equity Securities — D.
American Depositary Shares.” Our shareholders have shareholder rights. Australian law and our Constitution govern
shareholder rights. For a description of our shareholders’ rights, see “Item 10. Additional Information — B. Memorandum
and Articles of Association.”
ADS holders do not have the same voting rights as our shareholders. Shareholders are entitled to receive our notices of
general meetings and to attend and vote at our general meetings of shareholders. At a general meeting, every
shareholder present and entitled to vote has one vote on a show of hands. Every shareholder present (in person or by
proxy, attorney or representative) and entitled to vote has one vote per fully paid ordinary share on a poll. This is subject
to any other rights or restrictions that may be attached to any shares. ADS holders may exercise voting rights with
respect to the ordinary shares represented by the ADSs only in accordance with the provisions of the deposit agreement.
ADS holders may instruct the depositary to vote the ordinary shares underlying their ADSs. Otherwise, ADS holders are
not entitled to exercise their right to vote unless they surrender their ADSs and withdraw the ordinary shares underlying
their ADSs prior to both the ordinary share and ADS record dates for such meeting.
However, ADS holders may not have sufficient advance notice about the meeting to surrender their ADSs and withdraw
the shares. If we ask for ADS holders’ instructions, the depositary will notify registered holders of ADSs of the upcoming
vote and arrange to deliver our voting materials and form of notice to them. If we ask the depositary to solicit voting
instructions, the depositary will try, as far as practical, subject to Australian law and the provisions of the depositary
76
agreement, to vote the shares as ADS holders instruct. The depositary will not vote or attempt to exercise the right to
vote other than in accordance with the instructions of ADS holders. We cannot assure ADS holders that they will receive
the voting materials in time to ensure that they can instruct the depositary to vote their shares. In addition, there may be
other circumstances in which ADS holders may not be able to exercise voting rights.
ADS holders do not have the same rights to receive dividends or other distributions as our shareholders. Subject to any
special rights or restrictions attached to any shares, the directors may determine that a dividend will be payable on our
ordinary shares and fix the amount, the time for payment and the method for payment (although we have never declared
or paid any cash dividends on our ordinary shares and we do not anticipate paying any cash dividends in the foreseeable
future). Dividends may be paid on our ordinary shares of one class but not another and at different rates for different
classes. Dividends and other distributions payable to our shareholders with respect to our ordinary shares generally will
be payable directly to them. Any dividends or distributions payable with respect to ordinary shares represented by ADSs
will be paid to the depositary, which has agreed to pay to ADS holders the cash dividends or other distributions it or the
custodian receives on shares or other deposited securities, after deducting its fees and expenses. Before the depositary
makes a distribution to you in respect of your ADSs, any withholding taxes that must be paid will be deducted.
Additionally, if the exchange rate fluctuates during a time when the ADS depositary cannot convert the foreign currency,
you may lose some or all of the value of the distribution.
ADS holders will receive these distributions in proportion to the number of ordinary shares their ADSs represent. In
addition, there may be certain circumstances in which the depositary may not pay to ADS holders amounts distributed by
us as a dividend or distribution.
There are circumstances where it may be unlawful or impractical to make dividends or other distributions to the
holders of the ADSs.
The deposit agreement requires the depositary to convert foreign currency distributions it receives on deposited
ordinary shares into U.S. dollars and distribute the net U.S. dollars to ADS holders if it can do so on a reasonable basis
and transfer the money to the U.S. If it cannot make that conversion and transfer, the deposit agreement allows the
depositary to distribute the foreign currency only to those ADS holders to whom it is possible to do so. If a dividend or
other distribution is payable by us in Australian dollars, the depositary will hold the foreign currency it cannot convert for
the account of ADS holders who have not been paid. It will not invest the foreign currency and it will not be liable for any
interest. If the exchange rates fluctuate during a time when the depositary cannot convert the foreign currency, ADS
holders may lose some of the value of the dividend or other distribution. The depositary is not responsible if it decides
that it is unlawful or impractical to make a dividend or other distribution available to any ADS holders. This means that
ADS holders may not receive the dividends or other distributions we make on our ordinary shares or any value for them if
it is illegal or impractical for us to make them available to them.
There may be limited ability to bring an action against us or against our directors and executive officers, or to enforce
a judgment against us or them, because we are incorporated in Australia and certain of our directors and executive
officers reside outside of the U.S.
We are incorporated under the laws of Australia. Certain of our directors and executive officers are residents of countries
other than the U.S. and a portion of our and their assets are located outside of the U.S. As a result, it may not be possible
or practicable for owners or holders of ADSs to effect service of process within the U.S. upon such persons or to enforce
against us or them judgments obtained in U.S. courts predicated upon the civil liability provisions of the federal securities
laws of the U.S. Even if a plaintiff is successful in bringing such an action, there is doubt as to whether Australian courts
would enforce certain civil liabilities under U.S. securities laws in original actions or judgments of U.S. courts based upon
these civil liability provisions. In addition, awards of punitive damages in actions brought in the U.S. or elsewhere may be
unenforceable in Australia or elsewhere outside the U.S. An award for monetary damages under U.S. securities laws
would be considered punitive if it does not seek to compensate the claimant for loss or damage suffered and is intended
to punish the defendant. The enforceability of any judgment in Australia will depend on the particular facts of the case as
well as the laws and treaties in effect at the time. As a result, our holders of our ADSs may have more difficulty in
protecting their interests through actions against us, our management or our directors than would shareholders of a
corporation incorporated in a jurisdiction in the U.S. In addition, as a company incorporated in Australia, the provisions of
the Corporations Act 2001, regulate the circumstances in which shareholder derivative actions may be commenced,
which may be different to the circumstances for companies incorporated in the U.S.
The dual listing of our ordinary shares and the ADSs may adversely affect the liquidity and value of the ADSs.
Our ordinary shares are listed on the ASX and our ADSs are listed on Nasdaq. We cannot predict the effect of our dual
listing on the value of our ordinary shares and the ADSs. However, the dual listing of our ordinary shares and the ADSs
may dilute the liquidity of these securities in one or both markets and may adversely affect the development of an active
trading market for the ADSs in the U.S. The price of the ADSs could also be adversely affected by trading in our ordinary
shares on the ASX.
We are subject to risks associated with currency fluctuations, and changes in foreign currency exchange rates could
impact our results of operations.
Our ordinary shares are quoted in Australian dollars on the ASX and the ADSs are quoted in U.S. dollars. In the past year,
the Australian dollar has fluctuated against the U.S. dollar. As such, any significant change in the value of the Australian
77
dollar may have a negative effect on the value of the ADSs in U.S. dollars. In addition, if the Australian dollar weakens
against the U.S. dollar, then, if we decide to convert our Australian dollars into U.S. dollars for any business purpose,
appreciation of the U.S. dollar against the Australian dollar would have a negative effect on the U.S. dollar amount
available to us. While we engage in limited hedging transactions to manage our foreign exchange risk, these activities
may not be effective in limiting or eliminating foreign exchange losses. Consequently, appreciation or depreciation in the
value of the Australian dollar relative to the U.S. dollar would affect our financial results reported in U.S. dollar terms
without giving effect to any underlying change in our business or results of operations. As a result of such foreign
currency fluctuations, it could be more difficult to detect underlying trends in our business and results of operations.
As a foreign private issuer, we are permitted to follow, and we do follow, certain home country corporate governance
practices in lieu of certain Nasdaq requirements applicable to domestic issuers.
As a foreign private issuer listed on Nasdaq, we are permitted to follow, and we do follow, certain home country
corporate governance practices in lieu of certain Nasdaq practices. In particular, we follow home country law instead of
Nasdaq practice, and expect to continue to follow home country law instead of Nasdaq practice, regarding the following:
•We rely on an exemption from the requirement that our independent directors meet regularly in executive sessions.
The ASX Listing Rules and the Australian Corporations Act 2001 (Cth) do not require the independent directors of an
Australian company to have such executive sessions and, accordingly, we rely on this exemption.
•We rely on an exemption from the requirement that the responsibility for the appointment of the independent
registered public accounting firm be made by the audit committee. While our Audit and Risk Committee is directly
responsible for remuneration and oversight of the independent registered public accounting firm, the ultimate
responsibility for the appointment of the independent registered public accounting firm rests with our shareholders
in accordance with Australian law and our Constitution. In accordance with Rule 10A-3 of the U.S. Securities
Exchange Act of 1934, as amended ("Exchange Act") our Audit and Risk Committee is responsible for the annual
auditor engagement and if there is any proposed change to the independent registered public accounting firm, the
committee will make a recommendation to our board of directors, which would then be considered by our
shareholders at an annual meeting of shareholders.
•We rely on an exemption from the quorum requirements applicable to meetings of shareholders under Nasdaq rules.
Our Constitution provides that two shareholders present and entitled to vote on a resolution at the meeting shall
constitute a quorum for a general meeting. Nasdaq requires that an issuer provide for a quorum as specified in its
bylaws for any meeting of the holders of ordinary shares, which quorum may not be less than 33 1/3% of the
outstanding shares of an issuer’s voting ordinary shares. Accordingly, because applicable Australian law and rules
governing quorums at shareholder meetings differ from Nasdaq’s quorum requirements, we rely on this exemption.
•We rely on an exemption from the requirement prescribed by Nasdaq that issuers obtain shareholder approval prior
to the issuance of securities in connection with certain acquisitions, changes of controls or private placements of
securities, or the establishment or amendment of certain stock option, purchase or other compensation plans.
Applicable Australian law and rules differ from Nasdaq requirements, with the ASX Listing Rules providing generally
for the ability to seek prior shareholder approval in numerous circumstances, including (i) issuance of equity
securities exceeding 15% of our issued share capital in any 12 month period (but, in determining the available issue
limit, securities issued under an exception to the rule or with shareholder approval are not counted), (ii) issuance of
equity securities to related parties, certain substantial shareholders and their respective associates (as defined in
the ASX Listing Rules) and (iii) directors or their associates acquiring securities under an employee incentive plan.
Due to differences between Australian law and rules and the Nasdaq shareholder approval requirements, we rely on
this exemption.
As long as we remain subject to the rules of the ASX, we will be unable to access equity capital without shareholder
approval if such equity capital sales would result in an equity issuance above regulatory thresholds and,
consequently, we could be unable to obtain financing sufficient to sustain our business if we are unsuccessful in
soliciting requisite shareholder approvals.
Our ability to access equity capital is subject to ASX Listing Rules 7.1 and 7.4, which provides that a company must not,
without shareholder approval, issue or agree to issue any equity securities, or other securities with rights to conversion
to equity, if such issue of securities, when aggregated with securities issued by the company during the previous 12-
month period, would be an amount that would exceed 15% of the number of ordinary shares on issue at the
commencement of the 12-month period, subject to certain adjustments and permitted exceptions.
Our equity issuances are subject to limitations under ASX Listing Rule 7.1 as long as we continue to be listed on the ASX
and this constraint may prevent us from raising the sufficient equity capital needed to conduct our operations as planned
without shareholder approval.
As a foreign private issuer, we are permitted to file less information with the SEC than a company that files as a
domestic issuer.
As a foreign private issuer, we are exempt from certain rules under the Exchange Act that imposes disclosure
requirements as well as procedural requirements for proxy solicitations under Section 14 of the Exchange Act. In addition,
our officers, directors and principal shareholders are exempt from the reporting and “short-swing” profit recovery
78
provisions of Section 16 of the Exchange Act; although, absent an exemption from the SEC, and effective March 18,
2026, our officers and directors will be subject to the insider reporting obligations under Section 16(a) of the Exchange
Act, including the requirements to file Forms 3, 4 and 5, pursuant to the Holding Foreign Insiders Accountable Act
enacted on December 18, 2025. Moreover, we are not required to file periodic reports and financial statements with the
SEC as frequently or as promptly as a company that files as a domestic issuer whose securities are registered under the
Exchange Act. Under Australian law, we prepare financial statements on an annual and semi-annual basis, and we are not
required to prepare or file quarterly financial information.
For as long as we are a “foreign private issuer,” we intend to file our annual financial statements on Form 20-F and
furnish our semi-annual financial statements on Form 6-K to the SEC as long as we are subject to the reporting
requirements of Section 13(g) or 15(d) of the Exchange Act. However, the information we file or furnish is not the same
as the information that is required in periodic reports for U.S. domestic issuers. Accordingly, there may be less
information publicly available concerning us than there is for a company that files as a U.S. issuer.
We may lose our foreign private issuer status, which would then require us to comply with the Exchange Act’s
domestic reporting regime and cause us to incur additional legal, accounting and other expenses.
While we currently qualify as a foreign private issuer, we will be required to determine our status as a foreign private
issuer on an annual basis at the end of our second fiscal quarter. In order to maintain our current status as a foreign
private issuer, either (i) a majority of our ordinary shares must be either directly or indirectly owned of record by non-
residents of the U.S. or (ii) (a) a majority of our executive officers or directors must not be U.S. citizens or residents, (b)
more than 50 percent of our assets cannot be located in the U.S. and (c) our business must be administered principally
outside the U.S. If we lost this status, we would be required to comply with the Exchange Act reporting and other
requirements applicable to U.S. domestic issuers, which are more detailed and extensive than the requirements for
foreign private issuers. We may also be required to make changes in our corporate governance practices in accordance
with various SEC rules and Nasdaq listing standards. Further, we would be required to comply with U.S. GAAP, as
opposed to IFRS Accounting Standards, in the preparation and issuance of our financial statements for historical and
current periods. If we are required to comply with the reporting requirements applicable to a U.S. domestic issuer, the
regulatory and compliance costs to us may be higher than the cost we incur as a foreign private issuer. As a result, we
expect that a loss of foreign private issuer status would increase our legal and financial compliance costs.
If we fail to maintain proper internal controls, our ability to produce accurate financial statements or comply with
applicable regulations could be impaired.
The Sarbanes-Oxley Act ("Sarbanes-Oxley") requires our management to evaluate the effectiveness of our internal
control over financial reporting and disclose its conclusions, which includes identifying any material weaknesses in our
internal control over financial reporting. Beginning with this annual report that we are filing with the SEC, our
management is required to provide an annual report on internal control over financial reporting, and in the future, we
expect that our independent registered public accounting firm will be required to issue an annual report that addresses
the effectiveness of our internal control over financial reporting.
In order to maintain and improve the effectiveness of our disclosure controls and procedures and internal control over
financial reporting, we will need to expend significant resources and provide significant management oversight.
Implementing any appropriate changes to our internal controls may require specific compliance training of our directors
and employees, entail substantial costs in order to modify our existing accounting systems, take a significant period of
time to complete and divert management’s attention from other business concerns. These changes may not, however, be
effective in maintaining the adequacy of our internal control and preventing fraud.
If we are unable to conclude that we have effective internal control over financial reporting or, at the appropriate time, if
required, our independent auditors are unwilling or unable to provide us with an unqualified report on the effectiveness of
our internal control over financial reporting as required by Sarbanes-Oxley, investors may lose confidence in our
operating results, the price of the ADSs could decline and we may be subject to litigation or regulatory enforcement
actions. In addition, if we are unable to meet the requirements of Sarbanes-Oxley, we may not be able to remain listed on
Nasdaq.
We have identified a material weakness in our internal control over financial reporting. If we are unable to remediate
this material weakness, or if we identify additional material weaknesses in the future or otherwise fail to maintain an
effective system of internal controls, we may not be able to accurately or timely report our financial condition or
results of operations, which may adversely affect investor confidence in us and, as a result, the value of our ADSs.
We have identified a material weakness in our internal control over financial reporting as of December 31, 2025 as
disclosed in Item 15. Disclosure Controls and Procedures of this Annual Report on Form 20-F. A material weakness is a
deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable
possibility that a material misstatement of our annual or interim financial statements will not be prevented or detected on
a timely basis. As a result of the material weakness, management has concluded that our internal control over financial
reporting was not effective as of December 31, 2025. As a consequence of the material weakness, we have also
determined that our disclosure controls and procedures were not effective.
79
The material weakness related to segregation of duties, which have not been sufficiently established across the key
business and financial processes to maintain appropriate segregation of duties over certain manual and information
technology (“IT”) business controls. Segregation of duties is an internal control principle that helps prevent errors and
fraud by dividing tasks and responsibilities among different individuals. In our current control environment, due to the
size of our finance team, this segregation has not been adequately maintained. A consequence of the lack of segregation
of duties is a heightened risk of fraud or material misstatement where no appropriate mitigating controls are in place.
We have developed and begun to implement a remediation plan designed to improve our internal control over financial
reporting to remediate this material weakness. The remediation plan includes, and we have undertaken, redesigning roles
and access rights to enforce segregation of duties, implementing system-based workflow approvals, and enhancing
documentation of review controls.
As previously disclosed in Item 15 of the Annual Report on Form 20-F for the year ended December 31, 2024, we
identified a material weakness related to a lack of appropriately designed, implemented and documented procedures and
controls at both the entity-level and process-level to allow us to achieve complete, accurate and timely financial
reporting. We remediated this material weakness as of December 31, 2025, see “Item 15. Disclosure Controls and
Procedures.”
We cannot provide assurance that the measures we have taken to date, and measures we plan to implement, will be
sufficient to remediate the control deficiencies that led to the identified material weakness in our internal control over
financial reporting or that they will prevent or avoid potential future material weaknesses. If we are unable to successfully
remediate our existing or any future material weakness in our internal control over financial reporting, or identify any
additional material weaknesses in the future, or otherwise fail to maintain an effective system of internal controls, the
accuracy and timing of our financial reporting may be adversely affected, we may be unable to maintain compliance with
securities law requirements regarding timely filing of periodic reports in addition to applicable stock exchange listing
requirements, investors may lose confidence in our financial reporting, and the market price of our ADSs may decline as a
result.
We have incurred and will continue to incur significant increased costs as a result of operating as a company whose
ADSs are publicly traded in the U.S., and our management has devoted and will continue to be required to devote
substantial time to new compliance initiatives and corporate governance practices.
As a company whose ADSs are publicly traded in the U.S., we have incurred and will continue to incur significant legal,
accounting, insurance and other expenses that we did not previously incur. In addition, the Sarbanes-Oxley Act, Dodd-
Frank Wall Street Reform and Consumer Protection Act and related rules implemented by the SEC, have imposed various
requirements on public companies including requiring establishment and maintenance of effective disclosure and internal
controls. Our management and other personnel will need to devote a substantial amount of time to these compliance
initiatives, and we will need to add additional personnel and build our internal compliance infrastructure. Moreover, these
rules and regulations will increase our legal and financial compliance costs and will make some activities more time-
consuming and costly. These laws and regulations could also make it more difficult and expensive for us to attract and
retain qualified persons to serve on our board of directors, our board committees or as our executive officers.
Furthermore, if we are unable to satisfy our obligations as a public company in the U.S., we could be subject to delisting
of the ADSs, fines, sanctions and other regulatory action and potentially civil litigation.
We do not anticipate paying dividends in the foreseeable future.
We do not anticipate paying dividends in the foreseeable future. We currently intend to retain future earnings, if any, to
finance the development of our business. Dividends, if any, on our outstanding ordinary shares will be declared by and
subject to the discretion of our board of directors on the basis of our earnings, financial requirements and other relevant
factors, and subject to Australian law. As a result, a return on an investment in our ADS will only occur if the ADS price
appreciates. We cannot provide assurances that the ADSs will appreciate in value or even maintain the price at which
they are purchased. Investors may not realize a return on their investment in the ADSs and may even lose their entire
investment in the ADSs.
If securities or industry analysts do not publish research reports about our business, or if they issue an adverse
opinion about our business, the market price and trading volume of our ordinary shares or ADSs could decline.
The trading market for our ordinary shares and ADSs is influenced, in part, by the research and reports that securities or
industry analysts publish about us or our business. Securities and industry analysts may discontinue research on us, to
the extent such coverage currently exists, or in other cases, may never publish research on us. If no or too few securities
or industry analysts cover our Company, the trading price for our ordinary shares and the ADSs would likely be
negatively affected. If one or more of the analysts who cover us downgrade the ADSs or publish inaccurate or
unfavorable research about our business, the market price of the ordinary shares and ADSs would likely decline. If one or
more of these analysts cease coverage of us or fail to publish reports on us regularly, demand for the ordinary shares
and ADSs could decrease, which might cause our ordinary share and ADS prices and trading volumes to decline.
You may be subject to limitations on transfers of the ADSs.
The ADSs are transferable on the books of the depositary. However, the depositary may close its transfer books at any
time or from time to time when it deems expedient in connection with the performance of its duties. In addition, the
80
depositary may refuse to deliver, transfer or register transfers of ADSs generally when our books or the books of the
depositary are closed, or at any time if we or the depositary deems it advisable to do so because of any requirement of
law or of any government or governmental body, or under any provision of the deposit agreement, or for any other
reason.
Our Constitution and Australian laws and regulations applicable to us may adversely affect our ability to take actions
that could be beneficial to our shareholders and ADS holders.
As an Australian company listed on the ASX, we are subject to different corporate requirements than a corporation
organized under the laws of the U.S. Our Constitution, as well as the Australian Corporations Act 2001 (Cth) and ASX
Listing Rules, set forth various rights and obligations that are applicable to us as an Australian company listed on the
ASX. These requirements may operate differently than those of many U.S. companies. You should carefully review the
summary of these matters set forth under the section entitled “Item 10. Additional Information — B. Memorandum and
Articles of Association,” as well as our Constitution, which is included as an exhibit to this Annual Report, prior to
investing in the ADSs.
Australian takeover and foreign investment laws may discourage takeover offers being made for us or may
discourage the acquisition of a significant position in our ordinary shares or ADSs.
We are incorporated in Australia and are subject to the takeover and foreign investment laws of Australia. Among other
things, we are subject to the Australian Corporations Act 2001 (Cth) and Foreign Acquisitions and Takeovers Act.
Subject to a range of exceptions (including a takeover bid, scheme of arrangement or with shareholder approval), the
takeover provisions in the Australian Corporations Act 2001 (Cth) prohibit the acquisition of a direct or indirect interest in
our issued voting shares if the acquisition of that interest will lead to a person’s voting power in us increasing from 20%
or below to more than 20%, or increasing from a starting point that is above 20% and below 90%. Australian takeover and
foreign investment laws may discourage takeover offers being made for us or may discourage or prevent the acquisition
of a significant position in our ordinary shares. This may have the ancillary effect of entrenching our board of directors
and may limit the ability of our shareholders and ADS holders to obtain a premium from a control transaction.
We currently report our financial results under IFRS Accounting Standards, which differs in certain significant
respect from U.S. GAAP.
Currently we report our financial statements under IFRS Accounting Standards. There have been and there may in the
future be certain significant differences between IFRS Accounting Standards and U.S. GAAP, and those difference may
be material. As a result, our financial information and reported earnings for historical or future periods could be
significantly different if they were prepared in accordance with U.S. GAAP. In addition, we do not intend to provide a
reconciliation between IFRS Accounting Standards and U.S. GAAP unless it is required under applicable law. As a result,
you may not be able to meaningfully compare our financial statements under IFRS Accounting Standards with those
companies that prepare financial statements under U.S. GAAP.
There can be no assurance that we will not be a passive foreign investment company for any taxable year, which
could result in adverse U.S. federal income tax consequences to U.S. investors.
In general, a corporation organized outside the U.S. will be classified for U.S. federal tax purposes as a passive foreign
investment company ("PFIC"), for any taxable year in which either (i) 75% or more of its gross income consists of
“passive income,” or (ii) 50% or more of the value of its assets (generally determined on an average quarterly basis)
consists of assets that produce, or are held for the production of, passive income. For purposes of the above
calculations, a foreign corporation that owns (or is treated as owning) at least 25% by value of the shares of another
corporation is treated as if it held its proportionate share of the assets of that other corporation and received directly its
proportionate share of the income derived by that other corporation. “Passive income” generally includes dividends,
interest, rents, royalties and certain gains. Cash is a passive asset for these purposes.
Based on the expected nature and amount of our estimated gross income, the anticipated nature and estimated average
value of our gross assets, the anticipated cash needs of our group’s operations and the nature and extent of the active
businesses conducted by our “25% or greater” owned subsidiaries, we do not expect that we will be classified as a PFIC
in the current taxable year or for the foreseeable future. However, our PFIC status for any taxable year can be
determined only after the end of such year and will depend on the composition of our income and assets and the value of
our assets from time to time (which may be determined, in part, by reference to the market price of our ADSs or ordinary
shares, which could be volatile). Furthermore, the composition of our income and assets for the current and future
taxable years will be affected by how, and how quickly, we spend the cash we have on hand.
Accordingly, there can be no assurance that we will not be a PFIC for our current or any future taxable year. If we were a
PFIC for any taxable year during which a U.S. investor is treated as owning our ADSs or ordinary shares, the U.S. investor
generally would be subject to adverse U.S. federal income tax consequences, possibly including increased tax liability on
disposition gains and “excess distributions,” and additional reporting requirements. See “Item 10. Additional Information
— E. Taxation.”
Future changes to tax laws could materially adversely affect our company and reduce net returns to our
shareholders.
81
Our tax treatment is subject to the enactment of, or changes in, tax laws, regulations and treaties, or the interpretation
thereof, tax policy initiatives and reforms under consideration and the practices of tax authorities in jurisdictions in which
we operate, including those related to the Organization for Economic Co-Operation and Development’s Base Erosion and
Profit Shifting Project, the imposition of a minimum global effective rate for multinational businesses (Pillar Two) and
other initiatives. Such changes may include (but are not limited to) the taxation of operating income, investment income,
dividends received or (in the specific context of withholding tax) dividends paid. We are unable to predict what tax
reforms may be proposed or enacted in the future or what effect such changes would have on our business, but such
changes, to the extent they are brought into tax legislation, regulations, policies or practices, could affect our financial
position and overall or effective tax rates in the future in countries where we have operations, reduce post-tax returns to
our shareholders, and increase the complexity, burden and cost of tax compliance.
Tax authorities may disagree with our positions and conclusions regarding certain tax positions, or may apply
existing rules in an unforeseen manner, resulting in unanticipated costs, taxes or non-realization of expected
benefits.
We are subject to taxation in multiple jurisdictions. A tax authority may disagree with tax positions that we have taken,
which could result in increased tax liabilities. For example, although we believe we are compliant with applicable transfer
pricing requirements in various countries, a tax authority could challenge our allocation of income and the amounts paid
between our affiliated companies pursuant to our intercompany arrangements and transfer pricing policies. In the event a
tax authority assesses a deficiency, contesting such an assessment may be lengthy and costly and if we were
unsuccessful in disputing the assessment, the implications could increase our anticipated effective tax rate, where
applicable.
General Risk Factors
Unstable market and economic conditions may have serious adverse consequences on our business, financial
condition, results of operations and prospects and the trading price of our ordinary shares and the ADS.
Global credit and financial markets have experienced extreme disruptions over the past several years. Such disruptions
have resulted, and could in the future result, in diminished liquidity and credit availability, declines in consumer
confidence, declines in economic growth, increases in unemployment rates and uncertainty about economic stability. Our
general business strategy may be compromised by economic downturns, a volatile business environment and
unpredictable and unstable market conditions, such as pandemics or epidemics of infectious diseases, ongoing or future
wars or other geopolitical conflicts, imposition of tariffs or other trade restrictions, rising inflation, increasing interest
rates and slower economic growth or recession. If the equity and credit markets deteriorate, it may make any necessary
equity or debt financing more difficult to secure, more costly or more dilutive. Failure to secure any necessary financing
in a timely manner and on favorable terms could harm our growth strategy and financial performance and could require
us to delay or abandon plans with respect to our business, including clinical development plans. In addition, there is a risk
that one or more of our current service providers, manufacturers or other third parties with which we conduct business
may not survive difficult economic times, including ongoing or future wars or other geopolitical conflicts, and the
uncertainty associated with current worldwide economic conditions, which could directly affect our ability to attain our
operating goals on schedule and on budget.
Artificial intelligence-based platforms may present new risks and challenges to our business.
AI technologies may exacerbate existing risks, including risks associated with data privacy, cybersecurity, intellectual
property ("IP"), healthcare fraud and abuse, drug development and manufacturing, and risks to patients or human
subjects in clinical trials. AI also introduces new risks, due to the autonomous nature of the technology, which, in some
cases, may be deployed to perform tasks, inform decisions, automate decisions, and make predictions. AI may amplify
biased and discriminatory decision making, perform unreliably and malfunction, generate insights which are difficult to
interpret and explain, and cause direct harm to individuals or groups.
Regulators are proposing, adopting, and implementing new AI laws and regulations, including with respect to the
development of drugs and devices. We may be required to change our business practices and policies as a result of such
laws and regulations and may incur substantial compliance-related costs.
Regulators are also using existing laws and regulations to take enforcement actions related to the deployment of AI in
ways that result in non-compliance with current laws and regulations. If we fail to comply with AI laws and regulations,
we may be subject to sanctions, fines, and reputational damage, orders to stop certain processing of personal data,
orders to delete certain data or destroy AI algorithms derived from data collects, legal action on behalf of impacted
individuals or other enforcement or other actions. If we fail to take steps to protect our confidential data, trade secrets, IP
and personal data, we may be subject to legal, regulatory, financial, and reputational risks. AI technologies present
significant opportunities and risks to our business. Harnessing AI’s transformative potential may enable us to speed up
the discovery and development of new drugs and devices, optimize our manufacturing processes, and drive efficiencies.
Our failure to use AI technologies in a way that maintains trust, quality and control in our business activities and to
capitalize on opportunities presented by AI may also place us at a competitive disadvantage. Failure to address AI risks
will reduce our ability to deliver strategic objectives. Also, investments in AI may not realize the benefits that were
anticipated.
82
Business disruptions could seriously harm our future revenue and financial condition and increase our costs and
expenses.
Our operations and the operations of our suppliers, CROs, CMOs and clinical sites could be subject to earthquakes,
power shortages, telecommunications or infrastructure failures, cybersecurity incidents, physical security breaches,
water shortages, floods, hurricanes, typhoons, blizzards and other extreme weather conditions, fires, public health
pandemics or epidemics and other natural or manmade disasters or business interruptions, for which we are
predominantly self-insured. We rely on third-party manufacturers or suppliers to produce our Commercial Products and
our product candidates and on CROs and clinical sites to conduct our clinical trials, and do not have a redundant source
of supply for all components of our product candidates. Our ability to obtain sufficient supplies for our Commercial
Products and our product candidates could be disrupted if the operations of these suppliers were affected by a man-
made or natural disaster or other business interruption, and our ability to commence, conduct or complete our clinical
trials in a timely manner could be similarly adversely affected by any of the foregoing. The occurrence of any of these
business disruptions could seriously harm our operations and financial condition and increase our costs and expenses.
Global climate change, as well as increasing laws, regulation and litigation in the area of climate change, may have an
adverse effect on our results of operations, financial condition or liquidity.
There is widespread consensus in the scientific community that there is a long-term upward trend in global air and sea
temperatures that, along with shifting demographic trends in catastrophe exposed regions, has increased the severity
and frequency of severe weather events and other natural catastrophes, and is likely to further increase the average
economic value of expected losses in the future. Rising sea levels are also expected to increase the risk of coastal
flooding in many geographical areas. Extreme weather events can disrupt business continuity by negatively impacting
our infrastructure, systems and processes including, but not limited to, manufacturing and supply arrangements in
geographical locations exposed to severe weather events. In addition, global climate change could impair our ability to
predict the costs associated with future weather events. We cannot predict with certainty the frequency or severity of
hurricanes, tropical cyclones, wildfires or other natural catastrophes, and our risk assessments may not accurately reflect
shifting environmental and climate related risks. Unanticipated factors could lead to additional insured losses that exceed
our current estimates, resulting in disruptions to or adverse impacts on our business, the market or our third-party
collaborators.
ITEM 4.INFORMATION ON THE COMPANY
A.History and Development of the Company
Our legal name is Telix Pharmaceuticals Limited. Our company was incorporated under the laws of Australia in January
2017. In November 2017, we completed an initial public offering of our ordinary shares and the listing of our ordinary
shares on the ASX. Since November 13, 2024, we have been dual listed with our ADSs listed on the Nasdaq Global Select
Market, with each ADS representing one of our ordinary shares. JP Morgan Chase Bank, N.A., acts as depositary for the
ADSs.
Our corporate headquarters and registered offices are located at 55 Flemington Road, North Melbourne, Victoria, 3051,
Australia. Our reception telephone number is +61 3 9093 3855. Our agent for service of process in the U.S. is Telix
Pharmaceuticals (US) Inc., located at 11700 Exit 5 Pkwy, Suite 200, Fishers, Indiana 46037. Our website address is
www.telixpharma.com. All information we file with the SEC is available through the SEC’s Electronic Data Gathering,
Analysis and Retrieval system, which may be accessed through the SEC’s website at www.sec.gov.
The majority of our workforce is in the U.S., based out of our U.S. headquarters in Indianapolis, Indiana or our Dallas,
Texas office, our R&D and manufacturing facilities in Angleton, Texas and Los Angeles and Sacramento, California,
working remotely, or in one of our network of RLS Radiopharmacies that we acquired in January 2025. We also have R&D
and manufacturing facilities in Australia (North Melbourne, in completion phase), Belgium (Brussels South), Canada
(Vancouver) and Japan (Yokohama) and offices in Australia (Melbourne, Sydney and Brisbane), Belgium (Brussels and
Liège), Switzerland (Geneva), and Japan (Kyoto).
See “— B. Business Overview” (below) for a discussion of significant events and developments relating to our business
and “Item 5. Operating and Financial Review and Prospects — B. Liquidity and Capital Resources” for a discussion of our
capital expenditures.
B.Business Overview
Introduction
We are a global, commercial-stage biopharmaceutical company, focused on the development and commercialization of
therapeutic and diagnostic radiopharmaceuticals and associated medical technologies. We are committed to the
principles of precision oncology. By this we mean developing both therapeutic and diagnostic modalities for the benefit
of patients, an innovative precision medicine concept generally referred to as ‘theranostics’.
As an established leader and innovator in this field, Telix is differentiated by our deep expertise in radiopharmaceutical
drug development and commercialization, our innovative pipeline that spans the cancer care continuum, and our ability
83
to deliver patient outcomes globally. In order to achieve our mission and deliver on our strategy, Telix has structured the
business into five operating segments: Therapeutics (Tx), Precision Medicine (Px), International, MedTech and Telix
Manufacturing Solutions (TMS). We report financial results by the three reportable segments: Tx, Px (including
International and MedTech) and TMS. A brief overview is provided below.
•Therapeutics are at the core of the Telix portfolio, as we work to improve and extend patient life. We are currently
focused on developing targeted radionuclide therapies for urologic and neurologic oncology, and other solid
tumors.
•Precision Medicine is the commercial arm of Telix, focused on bringing diagnostic imaging solutions to market and
expanding into new geographies and indications. Precision medicine plays a fundamental role in managing patients
and delivering personalized therapeutic solutions. MedTech is advancing surgical solutions and digital products
that power Telix’s precision medicines and therapeutics. International is focused on “rest of world” (ex-U.S.)
commercial operations for the Europe, Middle East and Africa ("EMEA"), Asia Pacific ("APAC") and Latin America
regions.
•Telix Manufacturing Solutions is our global network of facilities designed to deliver patient doses worldwide. We
are investing in building manufacturing capacity, as well as improving the technology and processes to allow us to
deliver products at scale, including next generation alpha therapies. This investment in world-class infrastructure is
helping Telix to develop new products and secure critical supplies required to commercialize the future of cancer
treatments.
This commitment to end-to-end treatment differentiates Telix and is at the core of our radiopharmaceutical approach.
Australian Disclosure Requirements
Review of operations, likely developments and expected results; Business strategies and prospects for future years
A review of the Group’s operations for the financial year ended December 31, 2025, together with Telix’s business
strategies and prospects for future years, can be found in "Item 5. Operating and financial review and prospects" of this
Annual Report. Certain information regarding developments in operations in future years and expected results is
excluded, to the extent permitted by law, on the basis that such information relates to the impending developments or
matters in the course of negotiation and disclosure would likely result in unreasonable material prejudice to the Group.
Telix discloses its financial performance by operating segments. The Group’s operating segments represent components
of the Group that engage in distinct business activities. This provides the most meaningful insight into the nature and
financial outcomes of Telix’s activities and is consistent with the way in which the MD & CEO monitors and assesses
business performance and resource allocation decisions. Further details on Telix’s segment reporting can be found in
Note 3 of the Financial report.
Principal activities of the Company in the year under review
Telix’s principal activities during the year ended December 31, 2025 were directed to further advancing our standing as a
globally recognized theranostics company through executing on our strategy across four strategic pillars:
•Delivering our late-stage therapeutic pipeline: Development of TLX591-Tx (for prostate cancer), TLX250-Tx
(for kidney and other CAIX-expressing cancers), TLX101-Tx (for glioblastoma) and TLX66-Tx (for hematologic
cancers).
•Building the next generation of radiopharmaceuticals: Development of TLX592-Tx (TAT candidate for prostate
cancer), TLX252-Tx (TAT candidate for kidney and other CAIX-expressing cancers), TLX102-Tx (TAT candidate
for GBM and LMD), TLX300-Tx (TAT candidate for soft tissue sarcoma ("STS") and other PDGFRα expressing
cancers), TLX090-Tx (bone seeking candidate for bone metastases and pain palliation), and TLX400-Tx (FAP-
targeting therapeutic candidate with pan-cancer potential).
•Growing our industry leading precision medicine business: Development and commercialization of Gozellix and
Illuccix (focus on additional markets and indications), and development of TLX250-Px (Zircaix) and TLX101-Px
(Pixclara) and commercialization, if approved.
•Expanding our global infrastructure for product delivery: Grow manufacturing footprint and capabilities across
North America, Europe and the Asia Pacific region.
For more information on these four strategic pillars, refer to the "Our Strategy" section below.
Our Product Portfolio
Overview
Our portfolio includes both therapeutic and diagnostic radiopharmaceutical product candidates designed for use
throughout the continuum of the patient journey, from diagnosis and staging to treatment and ongoing care. We also
84
intend to use our therapeutic and diagnostic radiopharmaceutical product candidates in combination with one another,
as a theranostic treatment approach. Our clinical programs include several product candidates that are being evaluated
in Phase 1, Phase 2 and Phase 3 clinical trials with multiple expected upcoming data readouts and regulatory filings.
For most of our programs, particularly the prostate, kidney and brain programs, we have generated extensive clinical
data that we believe demonstrate the potential of our product candidates to offer meaningful benefits to patients. We
believe the targets and indications we are pursuing are well validated and are well suited for the delivery of therapeutic
and diagnostic targeted radiation. We believe that our use of imaging to select patients for therapy is also a
differentiated aspect of our commercial strategy. We believe that this precision medicine or theranostic approach may
increase the potential of our therapeutic development programs, as patients can be selected for therapy with greater
confidence that the drug target is sufficiently present to potentially confer therapeutic benefit. This may, in turn, lead to
more streamlined and efficient clinical trials, and enable improved patient outcomes.
A summary of our core development pipeline is illustrated below.
Therapeutic pipeline
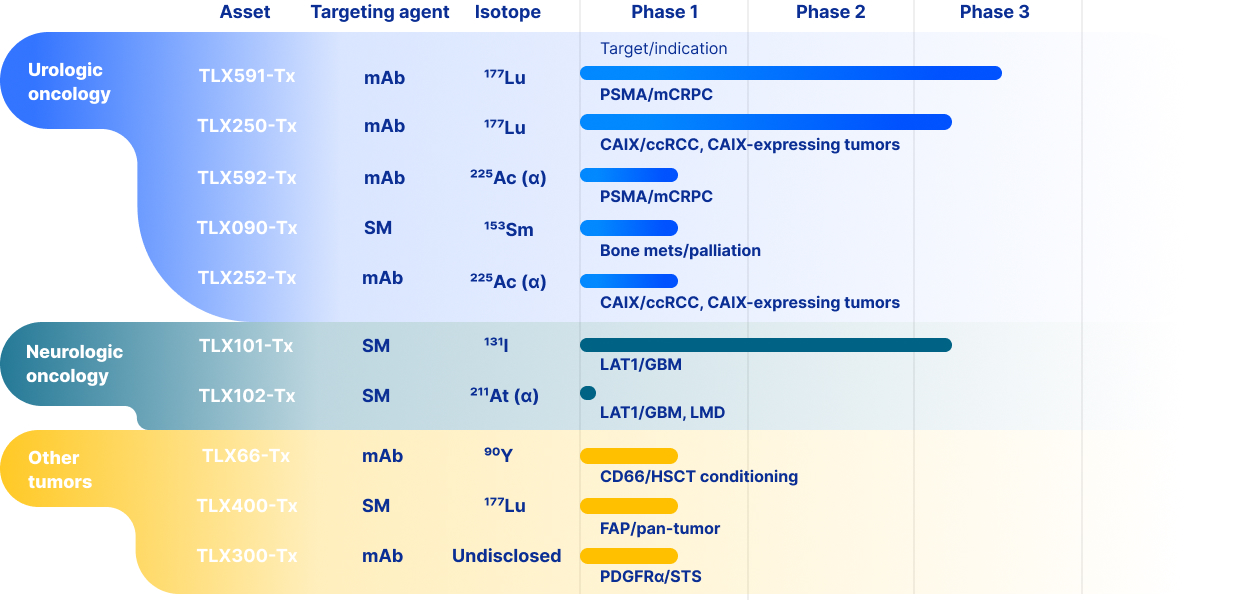
Precision medicine portfolio
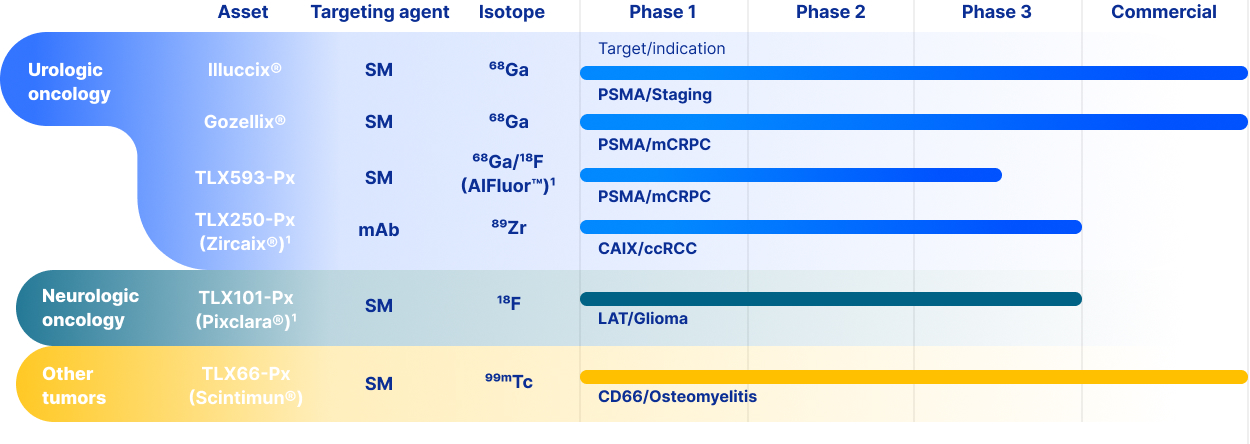
In addition to the development pipeline above, we are also exploring product and indication expansion opportunities with
our commercial and late-stage diagnostic portfolio through our lifecycle management programs. Lifecycle management
and product development includes two substantial prostate cancer indications for PSMA imaging, AlFluor™, a novel PET
radiochemistry solution based on 18F-aluminum fluoride ("AlF"), a staging indication for TLX250-Px, and an expansion into
brain metastases for TLX101-Px.
85
Prostate Cancer and PSMA
Our prostate cancer programs target PSMA, a well-validated protein target for the delivery of both therapeutic and
diagnostic radiopharmaceuticals that is highly expressed on prostate cancer cells with low expression on healthy cells.
We believe that our approach to PSMA therapy is unique because our lead product candidates, TLX591-Tx and TLX592-
Tx, use a monoclonal antibody carrier to target prostate cancer, whereas commercially available agents use a small
molecule peptide carrier. This approach allows for specific targeting of tumor tissue, differentiated pharmacokinetics and
excretion profiles, and prolonged treatment effect enabled by efficient irradiation of tumors. We are also developing a
small molecule PSMA therapeutic, TLX597-Tx, and a small molecule-based theranostic pair (RHN001-Tx for therapy and
RHN001-Dx for imaging, through our wholly-owned subsidiary Rhine Pharma), which are intended to enable access to
PSMA theranostics in select geographies.
TLX591-Tx
Our lead therapeutic product candidate TLX591-Tx (lutetium (Lu177) rosopatamab tetraxetan) is a lutetium-labelled rADC
that we believe has the potential to deliver improved patient outcomes with an efficient dosing regimen. The targeting
and pharmacology of TLX591-Tx differs significantly from PSMA-targeting small peptide molecules used in commercially
available compounds, and was designed for high internalization, long retention and to be highly selective for tumor-
expressed PSMA. This profile was designed with the goal of enabling a short, patient-friendly dosing regimen that
delivers a meaningful therapeutic index and low occurrence of off-target adverse events that are common with the
currently marketed PSMA-targeted small peptide molecules.
TLX591-Tx has demonstrated positive safety and efficacy signals following treatment of 242 patients across eight Phase
1 and Phase 2 clinical trials, including up to 42.3 months median survival in a single-arm Phase 2 clinical trial in 17
patients with mCRPC when delivered under a fractionated dosing regimen (Tagawa et al. Cancer. 2019). These studies
were not powered for or designed to demonstrate efficacy and results should be interpreted with caution. Efficacy
outcomes are presently being evaluated for statistical and clinical significance in a larger Phase 3 randomized controlled
trial.
ProstACT Global is a randomized, multinational, multicenter, open-label Phase 3 trial for the treatment of PSMA-positive
mCRPC patients in combination with the standard of care, compared to the standard of care alone. In August 2025, Part 1
of this trial, a dosimetry and safety lead-in portion, completed target enrollment of 30 patients. The results will be
presented to the FDA to obtain their agreement for the commencement of the randomized treatment expansion arm (Part
2), in the U.S. Part 2 already has regulatory approvals to commence in Australia, Canada, China, New Zealand, Singapore,
South Korea, Türkiye and the United Kingdom. We dosed the first patients in Part 2 in Australia in December 2025, and
are actively dosing patients at multiple clinical trial sites outside the U.S.
This is the first Phase 3 trial to evaluate TLX591-Tx in combination with the standard of care (androgen receptor pathway
inhibition or docetaxel) compared to the standard of care alone. The use of TLX591-Tx with current real-world standard
of care is intended to differentiate ProstACT Global from other PSMA-targeted trials and reflects our continued
innovation in prostate cancer care and commitment to patient outcomes.
TLX592-Tx
TLX592-Tx (225Ac-RADmAb), is our next generation prostate cancer therapy candidate for targeted alpha therapy ("TAT")
and is our first clinical program based on our proprietary RADmAb-engineered antibody technology. The engineered
antibody vector is designed for faster elimination from circulation than standard antibodies. Therefore, it is expected to
reduce bone marrow residence time, mitigating the risk of hematologic toxicities while retaining PSMA-mediated tumor
localization and exertion of cytotoxic activity. TLX592-Tx is designed to be cleared by the liver without exocrine uptake.
Prior to commencing therapeutic studies with the alpha emitting isotope 225Ac, we conducted the Phase 1 CUPID trial in
which we evaluated TLX592-Tx radiolabeled with the imaging isotope 64Cu, in patients with advanced prostate cancer.
We used 64Cu as a surrogate for 225Ac, since 64Cu is detectable by PET, whereas 225Ac is not. We investigated patients
with PSMA avid disease based on Illuccix imaging, across three mass dose levels, to assess the safety profile,
pharmacokinetics, biodistribution and dosimetry. Based on data from 11 evaluable patients, we observed accelerated
elimination from blood circulation compared to the original antibody used in TLX591-Tx, with similar on-target and off-
target biodistribution. There were no treatment-related adverse events observed in the trial.
We have received regulatory approval to commence the Phase 1 FIH therapeutic trial, AlphaPRO, in Australia, designed to
evaluate the safety profile of 225Ac-labelled TLX592-Tx.
TLX597-Tx
We are also developing a novel lutetium-labelled, small molecule-based, targeted radionuclide therapy candidate,
designated TLX597-Tx (177Lu-DOTA-HYNIC-panPSMA). This candidate is being developed to enable access to lutetium
therapy patients in select geographies, and is the subject of the ongoing Phase 2 OPTIMAL PSMA investigator-initiated
trial ("IIT"), which is expected to enroll 120 mCRPC patients in Australia.
86
RHN001-Tx
RHINO is an IIT sponsored by the Nuclear Medicine Research Infrastructure ("NuMeRI") at the University of Pretoria in
South Africa, exploring clinical utility of a theranostic pair derived from RHN001 (PSMA-GCK01), a PSMA-targeting small
molecule. RHN001 was developed through a collaboration between Telix and Heidelberg University Hospital ("UKHD"),
which resulted in pre-clinical validation and FIH evaluation of rhenium-188 (188Re) based radiopharmaceutical therapy and
technetium-99m (99mTc) based SPECT imaging (Cardinale et al. J Nucl Med. 2023).
This theranostic pair, designated RHN001-Tx (for therapy) and RHN001-Dx (for imaging), have the potential to enable
access to radiopharmaceuticals for patients in certain geographies where 177Lu availability may be limited. They can be
produced using compact, easily-transportable generators, potentially under practice of pharmacy as with other
generator-based radiopharmaceuticals, with imaging performed using widely available SPECT scanners.
The RHINO trial is a Phase 1/2a theranostic trial exploring the safety profile and dosimetry of both RHN001-Tx and
RHN001-Dx in patients with advanced prostate cancer. Its primary objective is to identify the recommended Phase 2
dose ("RP2D"), and schedule of administration for RHN001-Tx in men with progressive PSMA-positive prostate cancer
who have had adjuvant androgen deprivation therapy and/or taxane-based chemotherapy. Secondary objectives are to
assess the overall safety profile and the dosimetry of RHN001-Tx and RHN001-Dx in men with prostate cancer and
health-related quality of life.
The first cohort of patients received RHN001-Dx for safety and dosimetric analysis. The second cohort received
RHN001-Tx for safety and dosimetric analysis, while the third cohort (in progress) will receive escalating doses of
RHN001-Tx to identify the RP2D and schedule of administration for RHN001-Tx.
PSMA-PET imaging
Our prostate cancer portfolio also includes Illuccix and Gozellix, our commercially available 68Ga-labelled PSMA-PET
imaging agents. The “cold kit” format enables rapid radiolabeling at room temperature with high radiochemical purity and
production consistency, suited to the commercial and hospital radiopharmacy setting. Illuccix is approved in the U.S.,
Australia, Brazil, Canada, the United Kingdom and in 19 European Economic Area member states. Gozellix was approved
in the U.S. in March 2025 and the U.S. Centers for Medicare & Medicaid Services ("CMS") granted Transitional Pass-
Through ("TPT") payment status in September 2025. This designation enables separate reimbursement for Gozellix
under the Hospital Outpatient Prospective Payment System ("HOPPS"), taking effect from 1 October 2025 for three
years, marking a significant milestone in Telix’s U.S. commercial strategy. Approved indications for patients with prostate
cancer include staging of high-risk patients, identification of suspected recurrence, and selection for PSMA-directed
therapy (Illuccix only). In September we received a Prior Approval Supplement ("PAS") to update the U.S. Prescribing
Information for Illuccix. The Illuccix label now includes patient selection for RLT in the pre-taxane setting. During FY 2025,
our Precision Medicine business generated revenue of $621.9 million, primarily through sales of Illuccix and Gozellix.
We are exploring potential future utilization in additional indications for prostate cancer patients through our lifecycle
management program and growing our global PSMA-PET imaging presence through expansion into new geographies.
In June 2025, we launched a novel PET radiochemistry solution based on 18F-aluminum fluoride ("AlF"), named "AlFluor™".
The AlFluor platform technology enables flexible radiolabeling of PSMA with either AlF or 68Ga. It also has the potential to
be used with ligands targeting neuroendocrine tumors ("NETs") and FAP, as well as other novel imaging agents under
development by Telix and our strategic partners. Developed to combine the imaging benefits of 18F with the convenience
of 68Ga kit-based workflows, the AlFluor platform supports both centralized cyclotron manufacturing and distributed
“shake-and-inject” kit production. This flexibility allows a complementary product with the same targeting agent to be
labeled with either isotope, catering to clinical setting or physician preference. As part of AlFluor’s development, we
signed a strategic agreement with University Hospital Ghent and Ghent University for a novel [18F]AlF-PSMA-11 targeting
agent, which we have designated as TLX593-Px.
Bone Metastases and Pain Palliation
TLX090-Tx
TLX090-Tx (153Sm-DOTMP) is our novel kit-based bone-seeking targeted radiopharmaceutical product candidate that
uses a next generation chelating agent, DOTMP, to deliver a proprietary lower specific activity formulation of
Samarium-153 radioisotope, and is optimized for improved targeting, safety and clinical versatility. Importantly, TLX090-
Tx builds on FDA’s prior approval of the active moiety Samarium-153 (153Sm, used in Quadramet) while overcoming key
limitations of legacy formulations, such as skeletal saturation and calcium-binding risks.
153Sm is a beta-emitting radioisotope with a 46-hour half-life, and the chelating agent DOTMP selectively targets sites of
high bone mineral turnover, a known characteristic of bone metastases, while minimizing off-target migration. We believe
that TLX090-Tx may be administered as a single dose, multiple doses and higher dose regimens for pain management of
bone metastases and osteosarcoma therapy, including in pediatric patients. We believe that TLX090-Tx is highly aligned
with our existing therapeutic focus areas of prostate cancer, glioma and sarcoma.
In October 2025, we dosed the first patient in a Phase 1 clinical trial of TLX090-Tx called SOLACE (Samarium Optimized
for Long-lasting Analgesia in Cancerous End-stage bone pain). This open-label Phase 1 clinical trial is expected to enroll
up to 33 patients with advanced cancer that has metastasized to the bony skeleton and is designed to evaluate the
87
pharmacokinetics, dosimetry, safety, and pain palliation of TLX090-Tx. Data from SOLACE aims to establish clinical
comparability to legacy 153Sm treatments, which we believe may support a streamlined registration pathway as an
analgesic, paving the way for a much-needed, non-opioid solution for patients living with bone pain in the late stages of
advanced cancer. The novel cold-kit formulation and pharmacy-based distribution of TLX090-Tx may also aid in
overcoming barriers to treatment due to cost and supply chain limitations associated with legacy products.
Kidney Cancer and CAIX
Our target for kidney cancer is carbonic anhydrase IX ("CAIX"), a scientifically validated target in ccRCC, which is the
most prevalent and one of the most aggressive subtypes of kidney cancer. CAIX is a cell surface protein that is highly
expressed in ccRCC, and in many other solid tumors in the hypoxic tumor micro-environment. We believe this association
between hypoxia, disease progression, and therapeutic resistance underscores the relevance of this target. To target
CAIX, we use a monoclonal antibody, girentuximab, which is designed to have a high degree of selectivity and affinity for
the target. A single hepatically cleared agent is used for both imaging and therapy, with differing radioisotopes, to
minimize renal excretion, an advantage for assessing or treating primary disease. We believe the target profile and
properties of girentuximab make the ccRCC phenotype promising as the first therapeutic indication for TLX250-Tx, our
targeted radiation therapeutic product candidate.
TLX250-Tx
Our CAIX-targeting therapeutic candidate is TLX250-Tx (177Lu-DOTA-girentuximab), a rADC that we are developing for
the treatment of advanced metastatic kidney cancer.
We have had our ethics application approved in Australia and are presently initiating the LUTEON study, a global Phase
2/3 trial of of TLX250-Tx as a monotherapy in participants with recurrent ccRCC, who have progressive disease on or
after two and no more than three prior lines of therapy. The primary objective in Part 1 is to select the recommended
Phase 3 dose ("RP3D") to be administered in Part 2 from the two dose regimens administered in Part 1. The primary
objective in Part 2 is to compare the median progression free survival ("mPFS") of treatment with TLX250-Tx with that of
the SOC comparator as assessed by blinded independent central review.
TLX250-Tx is currently under investigation in the Phase 1b/2 STARLITE-1 investigator-initiated trial for kidney cancer,
where it is being evaluated in combination with immune checkpoint and tyrosine kinase inhibitors in up to 100 patients.
This single arm, Phase 2 study is evaluating the efficacy of TLX250-Tx in combination with nivolumab and cabozantinib
in patients with previously untreated ccRCC.
We believe the combined diagnostic and therapeutic potential of the girentuximab platform may also extend into other
cancers that significantly express CAIX, including certain Von Hippel Landau ("VHL") induced cancers, ovarian cancer,
triple-negative breast cancer and bladder cancer. We believe that proof-of concept imaging data in patients with triple-
negative breast cancer and bladder cancer supports future development of TLX250-Tx in these indications.
TLX252-Tx
TLX252-Tx (225Ac-DOTA-girentuximab) is a CAIX-targeting rADC alpha therapy candidate being developed as a potential
follow-on to the TLX250-Tx (beta) program.
TLX250-Px is used as an imaging surrogate for TLX252-Tx. The ZIRDOSE Phase 1 study characterized the biodistribution
and dosimetry of TLX250-Px in patients with ccRCC, enabling extrapolation to 225Ac (Merkx et al. EJNMMI. 2021).
Additionally, investigator-initiated trials demonstrated effective tumor targeting with TLX250-Px in triple-negative breast
cancer (Rousseau et al. EJNMMI. 2025) and non-muscle-invasive bladder cancer (Rousseau et al. Cancers. 2025),
indicating the potential for CAIX-targeted alpha therapy beyond ccRCC.
These findings, along with preclinical studies demonstrating efficacy for TLX252-Tx, support initiation of an
administered-activity escalation trial of TLX252-Tx for patients with advanced metastatic kidney cancer and other CAIX-
expressing tumors. Regulatory approval has been obtained in Australia for ALPHIX, a Phase 1 FIH study.
TLX250-Px
Our imaging candidate TLX250-Px is a PET diagnostic imaging agent that is under development to characterize
indeterminate renal masses as ccRCC or non-ccRCC in a non-invasive manner. The pivotal Phase 3 ZIRCON trial
evaluating TLX250-Px in 300 patients, of which 284 were evaluable, met all primary and secondary endpoints, including
showing 86% sensitivity and 87% specificity and a 93% positive-predictive value ("PPV"), for ccRCC across three
independent readers (Shuch et al. Lancet Oncol. 2024). We believe this demonstrated the ability of TLX250-Px to reliably
detect the clear cell phenotype and provide an accurate, non-invasive method for diagnosing ccRCC. Confidence
intervals exceeded expectations in all three readers, showing evidence of high accuracy and consistency of
interpretation.
We submitted a BLA for TLX250-Px to the FDA for regulatory approval in December 2023 for characterization of masses
as ccRCC. The BLA was granted on a rolling review process. We completed the BLA submission in May 2024, and in July
2024, the FDA declined to review the BLA and issued an RTF determination. The denial of acceptance for filing was
based on a filing concern related to demonstrating adequate sterility assurance during dispensing of TLX250-Px in the
88
radiopharmacy production environment. In December 2024 we resubmitted our BLA to the FDA for TLX250-Px, and the
FDA accepted our application in February 2025. In August 2025, the FDA issued a Complete Response Letter ("CRL")
citing deficiencies relating to the Chemistry, Manufacturing, and Controls ("CMC") package. The FDA requested
additional data to establish comparability between the drug product used in the ZIRCON Phase 3 clinical trial and the
scaled-up manufacturing process intended for commercial use. Additionally, the FDA documented notices of deficiency
(Form 483) issued to two third-party manufacturing and supply chain partners that required remediation prior to
resubmission. In December 2025 we participated in a Type A meeting with the FDA to align on plans to address CMC
deficiencies cited, and in January 2026 we participated in an additional Type A meeting to align on plans to address
comparability between the clinical drug product and that intended for commercial use. Following these meetings, Telix
believes it has alignment with the Agency on key resubmission aspects, and is working both internally and with third-
party vendors to implement these items for resubmission. While we believe that the planned remediation of the BLA for
TLX250-Px will meet FDA requirements, there can be no assurance that, once resubmitted, the FDA will accept our BLA
for review, or approve TLX250-Px. If approved, TLX250-Px would be the first targeted radiopharmaceutical imaging
agent for kidney cancer to be approved in the U.S.
In October 2025, TLX250-Px was included for the first time in joint guidelines from the Society of Nuclear Medicine and
Molecular Imaging ("SNMMI"), the European Association of Nuclear Medicine ("EANM") and The American College of
Nuclear Medicine ("ACNM") for molecular imaging of renal masses (Rowe et al. J Nucl Med. 2025). This is an important
development for Telix and will help raise awareness of this breakthrough precision diagnostic. Subject to regulatory
approval, endorsement by this expert global multidisciplinary panel will help to drive adoption and implementation into
clinical workflows, supporting clinical utility and enhancing decision-making to improve patient outcomes.
In November 2025, we announced new data from the ZIRCON-X study, which found that almost half of all patients
(48.6%) imaged with TLX250-Px PET/CT would undergo a change in clinical management, when compared with SOC
imaging. ZIRCON-X was a non-interventional, prospective, post-hoc study sponsored by Telix – using imaging data from
Telix’s parent pivotal Phase 3 ZIRCON study – that assessed the impact of TLX250-Px imaging on clinical decision-
making versus SOC contrast-enhanced diagnostic imaging in 294 patients with indeterminate renal masses ("IRMs").
As part of our commitment to provide access to medicine, we are running an expanded access program ("EAP") in the
U.S., named patient programs ("NPPs"), in Europe, and a special access scheme ("SAS") in Australia to allow access to
TLX250-Px outside of a clinical trial to patients for whom there are no comparable or satisfactory alternative options.
We also intend to conduct a label-expanding Phase 3 trial of TLX250-Px for the imaging of patients with metastatic
ccRCC. We believe TLX250-Px is a natural follow-on product to Illuccix as it is targeted at the same clinician users, the
urologist and urologic oncologist, and leverages our existing commercial infrastructure.
In June 2023, we dosed the first patient in the Phase 2 STARBURST trial of TLX250-Px exploring CAIX expression in
patients with a diverse range of solid tumors for potential therapeutic and diagnostic applications. This trial was designed
to identify new therapeutic indications for TLX250-Tx through the use of molecular imaging with TLX250-Px. In May
2025 the decision was taken to conclude the trial in the interests of utilizing clinical resources and patient time more
effectively - directing our focus towards theranostic studies that could offer both diagnostic insight and a novel
treatment option to the patients. We believe the integrated approach will yield greater clinical benefits for patients.
Glioma and LAT1/LAT2
Our targets for glioma are L-type amino acid transporters 1 and 2 ("LAT1" and "LAT2"), validated targets that are highly
expressed in several solid tumors, including malignancies of the central nervous system ("CNS"). We believe that the
LAT1 and LAT2 receptors, which are expressed on both sides of the blood-brain barrier, are suitable targets for the
delivery of radiation to both primary CNS malignancies and metastases from non-CNS cancers such as lung and breast
cancer. As such, we believe there are several potential indications for theranostic radiopharmaceuticals targeting LAT1
and LAT2.
TLX101-Tx
Our therapeutic product candidate, TLX101-Tx (iodofalan 131I), is a systemic therapy directed at the LAT1 receptor for the
treatment of glioblastoma. We are using a small molecule for this therapy due to the need to cross the blood-brain barrier
to reach its target. TLX101-Tx has received orphan drug designation in the U.S. and Europe for the treatment of glioma.
Orphan drug designation may not lead to a faster development or regulatory review or approval process and does not
increase the likelihood that TLX101-Tx will receive marketing approval.
We are evaluating TLX101-Tx in the front-line and recurrent disease settings where we have observed preliminary clinical
evidence of anti-tumor effect and disease stabilization. We completed the Phase 1/2 IPAX-1 trial of TLX101-Tx in
combination with external beam radiation therapy in patients with recurrent glioblastoma. IPAX-1 enrolled ten patients,
met its primary endpoint of safety and tolerability of TLX101-Tx and demonstrated preliminary efficacy data that
supports continued development (Pichler et al. Neuro-Oncology Advances. 2024). The Phase 1 IPAX-2 trial is designed to
enroll up to 15 patients to evaluate the safety of treatment of patients with newly diagnosed glioblastoma with TLX101-Tx
in combination with standard of care external beam radiation and chemotherapy. In IPAX-2, the second patient cohort
has completed and the third cohort is fully enrolled. As the primary goal of a Phase 1 study is to demonstrate safety and
tolerability criteria in a small patient population, these studies are not powered for or designed to demonstrate efficacy.
89
TLX101-Tx was also the subject of the investigator-led Phase 2 IPAX-Linz trial, in patients with recurrent glioblastoma,
which reported positive preliminary results in April 2025.
In July 2025, we received ethics approval in Australia to commence a Phase 3 trial of TLX101-Tx in patients with
recurrent glioblastoma, and in November 2025 we received health authority approval to commence the trial in Europe.
TLX101-Tx is the first radiopharmaceutical candidate for recurrent glioblastoma to enter Phase 3 development.
TLX102-Tx
TLX102-Tx (211At astato-L-phenylalanine, or 211At-APA) is a LAT1-targeting small molecule-based alpha therapy candidate
that we are developing as a potential complement to the TLX101-Tx and TLX101-Px programs. TLX102-Tx has
demonstrated pre-clinical proof-of-concept (Watabe et al. Oncotarget. 2020) and we believe that TLX102-Tx has the
potential for favorable efficacy and safety profile in future human clinical trials in patients with glioblastoma,
leptomeningeal disease, and multiple myeloma. Due to comparable target binding and molecular structure, we expect
that data from our existing LAT1 theranostic programs, TLX101-Px and TLX101-Tx, will complement and inform the
clinical and regulatory development strategy for TLX102-Tx. In August 2020, TLX102-Tx was granted orphan drug
designation from the FDA in the U.S. for the treatment of multiple myeloma and in November 2025, TLX102-Tx was
granted orphan drug designation from the FDA for the treatment of malignant gliomas. Orphan drug designation may not
lead to a faster development or regulatory review or approval process in multiple myeloma or glioma and does not
increase the likelihood that TLX102-Tx will receive marketing approval in either of these disease areas.
TLX101-Px
Our imaging candidate, TLX101-Px, also known as floretyrosine F18, is a PET diagnostic agent designed to image
cancerous lesions in the brain by targeting the LAT1 and LAT2 receptors. 18F-FET is widely used in many jurisdictions and
is recommended by the joint guidelines from the European Association of Nuclear Medicine ("EANM"), European
Association of Neuro-Oncology ("EANO"), Society of Nuclear Medicine and Molecular Imaging ("SNMMI"), Response
Assessment in Neuro-Oncology ("RANO"), The European Society for Pediatric Oncology and The Response Assessment
in Pediatric Neuro-Oncology for the characterization of recurrence in glioma patients (Galldiks et al. Lancet Oncol. 2025).
In June 2025, TLX101-Px was included in updated National Comprehensive Cancer Network® ("NCCN") Clinical Practice
Guidelines in Oncology ("NCCN Guidelines®") for Central Nervous System Cancers (V1.2025) for PET imaging of gliomas:
Delineation of tumor extent, diagnostic biopsy planning, radiotherapy planning for better visualization of tumor margins,
and differentiating tumor progression- and treatment-related changes per RANO/EANO/SNMMI guidelines. In October
2020, TLX101-Px was granted orphan drug designation by the FDA in the U.S. for the imaging of glioma. Orphan drug
designation may not lead to a faster development or regulatory review or approval process and does not increase the
likelihood that TLX101-Px will receive marketing approval.
In August 2024, we submitted a NDA to the FDA for TLX101-Px for the characterization of progressive or recurrent
glioma from treatment related changes in both adult and pediatric patients through the 505(b)(2) NDA regulatory
pathway. In October 2024, the FDA accepted the NDA, granted priority review and assigned a PDUFA goal date of April
26, 2025. In April 2025, the FDA issued a CRL citing the need for additional confirmatory clinical evidence to progress the
application. In September 2025 we reached agreement with the FDA regarding the required resubmission package,
which includes a retrospective analysis to supplement clinical data. We are currently finalizing our resubmission package
and will provide an update upon filing. There is no guarantee that the FDA will approve the NDA by the PDUFA goal date,
if at all. TLX101-Px was granted fast track designation by the FDA for this indication in April 2024. Fast track or orphan
drug designations may not lead to a faster development or regulatory review or approval process, and do not increase
the likelihood that TLX101-Px will receive marketing authorization.
In February 2026, we submitted a MAA for TLX101-Px in a selection of European countries, with France acting as RMS.
The French National Agency for Medicines and Health Products Safety, ANSM, in its capacity as RMS is responsible for
coordinating and leading the scientific evaluation of the dossier, in collaboration with the Concerned Member States.
There can be no assurance that the MAA will be validated, or that marketing authorizations for TLX101‑Px will ultimately
be granted.
As part of our commitment to provide access to medicine, we are running an EAP in the U.S. to allow access to TLX101-
Px outside of a clinical trial to patients for whom there are no comparable or satisfactory alternative options.
We also intend to conduct a label-expanding Phase 3 trial of TLX101-Px for the imaging of patients with brain metastases
from non-brain cancers, including lung and breast cancer.
Other Solid Tumors and Hematologic Oncology
Soft Tissue Sarcoma and PDGFRα: TLX300-Tx and TLX300-Px
Our product candidates TLX300-Tx and TLX300-Px employ antibody-directed targeted radiation for both therapeutic
and patient selection applications against platelet-derived growth factor receptor alpha ("PDGFRα"); activation of
PDGFRα is associated with cancer proliferation, metastasis, invasion, and angiogenesis. Eli Lilly Kinsale Limited provided
us with a license for olaratumab, a naked antibody that was formerly marketed as Lartruvo. We re-purposed olaratumab
as a radiopharmaceutical product candidate.
90
In April 2025, we dosed the first patient in ZOLAR, a first-in-human Phase 1 proof-of-concept targeting and
biodistribution trial using TLX300-Px. We intend to develop the therapeutic application of TLX300-Tx for the treatment of
advanced soft tissue sarcoma ("STS") and other PDGFRα expressing cancers. We have not yet determined the specific
therapeutic isotope that we will use in therapeutic trials.
TLX300-Px (89Zr-DFOsq-olaratumab, including our proprietary DFO-squaramide chelator) is an imaging candidate that
we are developing for use with TLX300-Tx as a theranostic pair.
Hematologic Oncology and CD66
In hematologic oncology and bone marrow conditioning ("BMC"), we are exploring the potential utility of targeted
radiation to ablate bone marrow as part of a pre-conditioning regimen for bone marrow transplantation, novel stem cell
therapies and gene therapies, each of which requires conditioning prior to treatment. The standard of care involves using
highly toxic chemo-ablation techniques that require long hospitalization times and significant treatment-related morbidity
and mortality risks, which considerably limit patient access to these therapeutic interventions. We believe that a safe,
durable and short internment treatment could be transformative to many facets of cancer and autoimmune disease
treatments that require BMC.
TLX66-Tx
Our antibody-based product candidate TLX66-Tx (90Y-DTPA-besilesomab) is designed to target cluster of differentiation
66 ("CD66"), a well-validated leukocyte and neutrophil target. TLX66-Tx has been evaluated as a therapeutic bone
marrow conditioning agent in approximately 80 patients with results that support continued development, both as a
monotherapy and in combination with low dose chemotherapy conditioning regimes (Orchard et al. Bone Marrow
Transplantation. 2024). In June 2025, the first patient was dosed in a Phase 2 IIT in pediatric high-risk leukemia. We also
plan to evaluate TLX66-Tx in a Phase 2 clinical trial as a BMC agent in patients with acute myeloid leukemia who are not
suitable for conventional BMC regimes.
In March 2022, TLX66-Tx was granted orphan drug designation by the FDA in the U.S. as a conditioning treatment prior
to hematopoietic stem cell transplant ("HSCT"). TLX66-Tx was granted orphan drug designation in Europe in October
2019. Orphan drug designation may not lead to a faster development or regulatory review or approval process and does
not increase the likelihood that TLX66-Tx will receive marketing approval.
We believe that the imaging application of besilesomab could support patient selection for TLX66-Tx by informing
healthcare providers whether sufficient activity will be absorbed by a patient’s bone marrow. TLX66-Px, an imaging
application of besilesomab, has already been commercialized and is an approved product (marketed as Scintimun) for
imaging osteomyelitis (bone infection) in 32 European countries and Mexico. Scintimun was previously manufactured and
distributed by Curium Pharma through an out-license from Telix via the acquisition of TheraPharm in 2020. Following a
strategic review of the asset, we brought sales and marketing in-house during 2025, with plans to significantly augment
commercial distribution and indication expansion. TLX66-Px has not received marketing approval in the U.S.
Pan-Cancer, CAIX and FAP
Our pan-cancer programs target CAIX and fibroblast activation protein ("FAP"), two well-validated targets with pan-
cancer potential. This complementary approach targeting CAIX and fibrosis is a potential “double hit” at the tumor micro-
environment, with potential to provide a synergistic benefit when combined with other systemic treatments such as
immuno-therapies.
We have observed encouraging preliminary clinical data in bladder and breast cancers with our CAIX-targeting TLX250
platform, and in March 2025, we completed a transaction to expand our theranostic pipeline with a portfolio of next
generation FAP-targeting assets. Our FAP development program is exploring potential in a range of solid tumors, as
many cancers are known to express this target either in the tumor micro-environment, like breast cancer, or directly on
malignant lesions, like sarcomas. Our lead therapeutic candidate, TLX400-Tx, has been administered in approximately
150 patients in a compassionate use setting covering multiple tumor types.
Operations and Manufacturing Activities
Our corporate headquarters are located in Melbourne, Australia. The majority of our workforce is in the U.S., based out of
our U.S. headquarters in Indianapolis, Indiana, or our Dallas, Texas office, our R&D facilities in Angleton, Texas and Los
Angeles and Sacramento, California, working remotely, or in one of our network of radiopharmacies that we acquired in
January 2025 as part of our acquisition of RLS. We also have facilities in Australia (Melbourne, Sydney and Brisbane),
Belgium (Brussels and Liège), Switzerland (Geneva), Japan (Kyoto and Yokohama) and Canada (Vancouver). We are
investing significantly to build a world-class vertically integrated supply chain, superior manufacturing and distribution
capabilities, and the ability to deliver radiopharmaceuticals to all major global markets.
We believe the impact of our investment into supply chain, manufacturing, distribution, and commercial capabilities to
date is clearly demonstrated through the successful commercial launch of Illuccix. Leveraging our extensive network of
partners, we have expanded manufacturing capabilities to support the scale-up of commercial sales of Illuccix and, since
2025, Gozellix. Furthermore, our widespread distribution network, encompassing over 225 radiopharmacies across the
U.S., is designed to ensure flexibility and reliability in delivering Illuccix and Gozellix imaging doses to patients.
91
We continue to invest to strengthen our vertically integrated supply chain and manufacturing model. In 2023 we opened
our TMS facility located in Brussels South, Belgium. At approximately 30,000 square feet, it is one of the largest
radiopharmaceutical production facilities in Europe, with nine GMP lines, clean rooms, a radiopharmacy, and two
cyclotrons. We expect this facility to deliver significant flexibility and reliable supply for our growing commercial
production requirements. It also serves as a vital hub for research and development, specifically in manufacturing scale-
up and production of next generation radiopharmaceuticals, including both alpha-emitters and beta-emitters. In May
2025, TMS Brussels South received notice of GMP accreditation for Illuccix, and delivered first GMP grade commercial
radiopharmaceutical doses in June 2025.
In December 2022, we acquired Optimal Tracers, a Sacramento-based company that provides radiochemistry process
development services and research tracers for use in clinical trials. The acquisition expanded our translational
radiochemistry capability and established a U.S.-based laboratory (now known as TMS Sacramento) and production
footprint for manufacturing doses of radiopharmaceutical to support clinical trials.
In April 2024, we acquired IsoTherapeutics, which is enabling us to internalize select aspects of our development
programs, with the goal of reducing cost and time to achieve technical milestones.
In April 2024, we acquired ARTMS, further enhancing the vertical integration of our supply chain and manufacturing, with
the goal of facilitating broader patient access to therapeutic and diagnostic radiopharmaceuticals through ARTMS’ high-
yield production techniques. In March 2025, ARTMS' drug master file ("DMF"), for gallium-68 production using its
QUANTM Irradiation System® ("QIS®") cyclotron technology was referenced for the first time with a FDA-approved
product, with Gozellix. This approval enables radiopharmacies and hospitals using QIS and associated targets to produce
multi-Curies of 68Ga for use with Gozellix. We believe that the expanded distribution radius and increased dose
production capacity, compared to existing gallium-based PET tracers, will facilitate broader and more equitable access.
In January 2026, ARTMS' DMF for gallium-68 production using QIS was referenced with Illuccix. In addition, use of
ARTMS’ technology is being expanded internationally including at Telix cyclotron facilities. This includes TMS Brussels
South, where QIS was installed on both cyclotrons during 2025, with zirconium-89 and gallium-68 subsequently
produced.
In January 2025, we acquired RLS (USA) Inc., the only Joint Commission-accredited U.S. radiopharmacy network
distributing PET, SPECT and therapeutic radiopharmaceuticals. The RLS acquisition – including over 30 radiopharmacies
across the U.S. – augments our existing distribution network for last-mile delivery and provides expansionary space to
build out a radiometal production network to meet future demand for radiopharmaceuticals. The process to install
cyclotrons at selected RLS radiopharmacy locations is underway.
In March 2025, we announced the establishment of a joint venture with R2PHARMA, structured as Telix Innovations Brazil
Ltda. (the "JV"), to commercialize and distribute our therapeutic and diagnostic radiopharmaceutical products in Brazil,
The JV further strengthens the partnership established in 2019, with a commitment to jointly bring to market innovative
and first-in-class therapeutic radiopharmaceuticals and imaging agents in Brazil. The JV is expected to hold an exclusive
license to commercialize and distribute Illuccix, as well as future product candidates, once all required regulatory,
licensing, and governmental authorizations are obtained. Until such authorizations are granted, Illuccix continues to be
distributed in Brazil by one of the JV’s affiliated companies within the R2PHARMA Group.
In June 2025, we established TMS Yokohama in Japan, Telix’s first cyclotron facility in the APAC region. TMS Yokohama
will serve as a hub for commercial and clinical supply, and future research and development. It expands Telix’s global
production network which includes in-house and partner facilities. Originally opened in 2018, the site comprises a
cyclotron and multiple production hot cells and was designed and built by JFE Engineering ("JFE"), as the Contract
Manufacturing Organization ("CMO"), for TLX250-Px in Japan and China, including for the ZIRCON-CP study. We believe
that taking over the lease and operational management of this facility will provide greater control over existing clinical
supply with the possibility to expand production to other Telix investigational and future commercial products in the
region, including Illuccix, and TLX101-Px for Greater Tokyo, and TLX591-Tx for the APAC region, if marketing
authorization of the product candidates is granted. Further, we plan to install ARTMS' QIS cyclotron technology, which
we believe will facilitate standardized, high-efficiency and cost-effective production of zirconium-89 supporting TLX250-
Px production.
During 2025, we progressed the construction of a new facility in Melbourne, Australia, to be known as TMS North
Melbourne, for early-phase clinical research and radiopharmaceutical production, to support APAC translational research
and clinical trials. In September 2025, both TMS North Melbourne and TMS Yokohama were granted radiation licenses
for a broad range of clinically and commercially important medical isotopes.
Our Opportunity
The global radiopharmaceutical industry is undergoing a period of transformative growth with theranostics emerging as a
key pillar in the armamentarium of oncology treatment. We believe that with increasing integration of nuclear medicine
and traditional oncology clinical practice, radiopharmaceuticals will become a core component of the multi-disciplinary
approach to cancer treatment with a proportionate benefit to patients.
Our therapeutic radiopharmaceutical platform harnesses the power of radioactive isotopes combined with multi-platform
targeting agents to deliver targeted radiation directly to the tumor site. These therapeutic product candidates have the
potential to be stand-alone treatments or as complements to existing treatment modalities to address areas of high
92
unmet medical need. Due to our expertise in the multiple components of radiopharmaceuticals we believe we are able to
create theranostics in an “agnostic” manner, pairing the right delivery mechanism with the right isotope most likely to be
suited for the tumor being treated.
We intend to pair each therapeutic with a diagnostic imaging agent, this underpins the “theranostic” approach whereby
two conjugates are used to target the same cell-surface receptor, one for detection, localization or staging, and the other
for selective destruction of target cancer cells. When used in tandem to plan and execute treatment, and then to assess
response and monitor for progression, we believe this approach allows the delivery of truly personalized therapy to
patients.
Our Strategy
Our strategy is to launch innovative imaging agents in our core disease areas, while financing the development of
therapeutic product candidates, including next generation radiopharmaceuticals. This strategy is underpinned by a
vertically integrated approach to supply and manufacturing, and is supported by a first-class commercial organization
ensuring global patient access to our products and product candidates, if approved.
Our strategy is designed to deliver on our purpose and mission and reflects the evolution of our business into a global,
multi-product commercial-stage company with a deep theranostic pipeline. This approach, we believe, will deliver
benefits to patients and returns to shareholders over the near and long-term.
The four strategic pillars central to achieving our mission are:
1. Deliver our late-stage therapeutic pipeline
We aim to build both breadth and depth in oncology and to address areas of significant unmet medical need, both for
large oncology indications such as prostate cancer and kidney cancer, as well as rare oncology indications such as
glioma. This is based on a target selection process that is aligned with our expertise in radiation biology.
In August 2025, we completed target enrollment in Part 1 of the Phase 3 ProstACT Global study of TLX591-Tx (lutetium
(Lu177) rosopatamab tetraxetan), our lead rADC candidate for advanced prostate cancer. In December 2025, we dosed
first patients in Part 2 (randomization) in Australia with sites also recruiting in Canada and New Zealand. The study is also
approved to commence in China, Japan (Part 1 only), Singapore, South Korea, Türkiye and the United Kingdom. This
multinational Phase 3 study is to investigate and evaluate the potential benefits and risks associated with TLX591-Tx,
when administered together with SOC and compared to SOC alone.
We are also advancing TLX250-Tx and TLX101-Tx into late-stage clinical trials for the treatment of kidney cancer and
glioblastoma, respectively. In 2025, we received regulatory approval in Australia and the European Union to commence
the Phase 3 IPAX-BrIGHT pivotal trial of TLX101-Tx with sites open for enrollment, and received regulatory approval in
Australia to commence LUTEON, a global Phase 2/3 trial of TLX250-Tx. We believe that each of our product candidates
is currently the most advanced systemic radiotherapy in its respective indication.
2. Build the next generation of radiopharmaceuticals
We have established a track-record in identifying validated clinical product candidates that can be optimized as
radiopharmaceutical therapies to develop them through to commercial products. We are leveraging this capability to
expand our pipeline with next generation radiopharmaceuticals, particularly targeted alpha-emitting therapies, through
business development, as well as internal R&D programs and collaborations. Through our existing clinical programs and
dedicated research facilities in the U.S., Australia and Europe, we are focused on the development of alpha therapy
candidates as a future pipeline expansion opportunity, and on building supply and manufacturing capabilities required to
support an eventual commercial launch.
We are advancing TLX592-Tx, TLX252-Tx and TLX102-Tx, our next generation (alpha) therapy candidates for prostate,
kidney and brain cancer into clinical trials. In 2025, we received regulatory approval to commence Phase 1, first-in-
human studies of TLX592-Tx and TLX252-Tx. During the year, we dosed first patients in a Phase 1, first-in-human
imaging study of TLX300-Px, aiming to demonstrate proof of concept for therapy in soft tissue sarcoma ("STS") and
other platelet derived growth factor receptor alpha ("PDGFRα") positive tumors. In 2025, we dosed first patients in the
Phase 1 SOLACE study of TLX090-Tx, Telix's therapy candidate for pain palliation of osteoblastic bone metastases, and
also acquired a suite of clinically validated theranostic candidates (including TLX400-Tx) targeting fibroblast activation
protein ("FAP"), widely considered to be one of the most promising targets for both cancer imaging and therapy.
In January 2025, we acquired a pipeline of next generation therapeutic candidates, a proprietary novel biologics
technology platform, and a protein engineering and discovery research facility based in Los Angeles, California, from
antibody engineering company ImaginAb, Inc. ("ImaginAb"), to enhance existing innovation capabilities. This provides us
with a pipeline of early-stage drug candidates against high-value targets including delta-like ligand 3 ("DLL3"), and
integrin αvβ6, as well as several other novel targets in discovery stage. We believe these next generation drug
candidates fit synergistically with our therapeutics pipeline, enabling expansion to future therapy areas with unmet
clinical need. The acquired intellectual property utilizes small engineered antibody formats that enable highly specific
cancer targeting, combined with fast tumor uptake and blood clearance. This technology has the potential to be highly
effective for imaging and treating tumors with a broad range of radioisotopes, with alpha emitters of particular interest.
93
In March 2025, we announced a validated breakthrough generator technology for the production of the alpha-emitting
isotope, lead-212 (212Pb) and successfully completed first production. The new generator technology, being developed
internally by our IsoTherapeutics team, produces 212Pb via a Thorium-228 (228Th) emanation source, and significantly
increases the amount of radioactivity, yield and shelf life compared to currently available 212Pb generators. The
production footprint has been designed to be deployed throughout Telix’s (and select partner) manufacturing and
distribution networks, including RLS Radiopharmacies.
3. Grow our industry-leading Precision Medicine business
Leveraging the success of our first commercial product, Illuccix, we intend to broaden our commercial footprint in
precision medicine by:
•Expanding Illuccix and Gozellix into new indications and geographies and the continued successful launch of
Gozellix in 2026.
•Obtaining marketing authorization for TLX250-Px, which will expand our urology portfolio with potential to become
the first-in-class targeted radiopharmaceutical imaging agent for ccRCC.
•Obtaining marketing authorization for TLX101-Px for the imaging of glioma.
4. Expand our global infrastructure for product delivery
To develop and deliver radiopharmaceuticals, which have a relatively short shelf life due to their unique properties,
supply chain, manufacturing and logistics are vitally important. TMS is a global network of facilities, where our continued
focus and investment in infrastructure and technologies is equipping Telix to deliver patient doses worldwide.
Telix has made a number of strategic acquisitions to grow our manufacturing capability and footprint. Going forward,
TMS will be focused on activities including:
•Preparing selected RLS Radiopharmacies sites for cyclotron installation with ARTMS QIS technology, to increase
efficiency and improve yields when producing radioisotopes, and establish a next generation radiometal production
network.
•Expanding the deployment of the ARTMS QIS technology including at our state-of-the-art TMS facility in Brussels
South, Belgium – one of the largest of its kind in Europe, and TMS Yokohama.
•Increasing GMP production at TMS Brussels South.
•Completing the fit-out at TMS North Melbourne and scaling up of operations at TMS Yokohama.
Our Theranostic Approach
Our approach enables us to design and develop product candidates to deliver targeted radiation to cancer cells,
regardless of where the cancer is in the body, via a systemic radioactive infusion. We aim to use imaging and therapy
together to “see and treat” cancer. We refer to this approach as theranostic, which we believe is a powerful way to tackle
unmet need in cancer and rare diseases.
We believe that our ability to harness the power of targeted radiation throughout the patient journey to enhance patient
outcomes is a key differentiator.
Targeted Radiation Overview
We are developing targeted radiation across the continuum from diagnosis and staging to treatment, both as stand-alone
and combination therapies.
Many existing cancer therapies are non-selective and as a result can act against healthy tissue and vital organs while
treating disease. Existing external beam radiation therapy ("EBRT"), approaches are effective but typically only deliver
localized treatment and cause damage to surrounding tissue. Localized therapeutic approaches rely on the treating
physician making assumptions about the extent of disease and can result in imprecise application of treatment.
Treatments that miss small amounts of cancer cells can lead to a recurrence of the cancer or disease.
Targeted radiation uses a radioactive isotope as a payload that is attached to a targeting agent, such as a small molecule
or antibody, with an affinity for specific biomarkers found on the surface of cancerous or diseased cells. Depending on
the choice of radioisotope payload, these target agents can deliver either imaging or therapy.
The targeted radiation drug or antibody is administered into the bloodstream and circulates throughout the body. Once
administered, the targeted radiation seeks cancerous or diseased cells, including primary tumors and small metastases
(where the cancer has spread), upon which it is designed to bind selectively to its target. Some radioactive isotopes have
physical properties that may be used to image cancer or rare diseases, for diagnosis and staging purposes. Higher dose
radiation with different alpha- and beta-emitting radioisotopes can be used as therapies to kill cancerous or diseased
cells, by damaging their DNA.
94
The Targeting Agent
The targeting agent guides the radiation payload to the targeted cancer cells. The agent is designed to be cancer-
specific due to selective affinity for tumor targets that are prevalent in tumors but not healthy tissues. The targeting
agents can be either an antibody, engineered antibody fragment, peptide or small molecule, and the choice of targeting
agent can impact the other properties of the drug, including:
•Pharmacokinetics: Peptides and small molecules have a short circulation time (several hours) and therefore require
a higher dose of radiation payload to sufficiently irradiate the tumor in therapeutic contexts, which comes at the
expense of a resulting higher radiation exposure to the kidney. Antibodies have a longer circulation time (several
days), are cleared though the liver and are lost slowly, which can transiently impact the levels of blood cells but
results in higher amounts of radiation payload in tumors to maximize the therapeutic effect. The calculations and
study required to determine the optimal dose of radiation to be delivered for maximum therapeutic effect with an
acceptable safety profile are referred to as "dosimetry".
•Binding and cancer specificity: Antibodies have evolved in the immune system to be highly selective and, as a
well-known class of agents, can be generated to be highly specific to their target. Small molecules and peptides are
not as predictable as a delivery platform, however they can be engineered for high selectivity and affinity; their
metabolism properties and off-target toxicity are unique to each molecule.
•Internalization and residualization in the tumor: Once bound to their biological targets, targeting agents can be
taken up by cancer cells through a process called "internalization". Peptides tend to be returned to the blood or
otherwise degraded relatively quickly after internalization. By contrast, antibodies tend to be retained within cancer
cells and, with their sustained presence in the blood, tend to accumulate or ‘residualize’ their radiation payload over
time which can favor the localization of higher amounts of radiation to the tumor than peptides or small molecules.
The slow excretion of antibodies and their ability to highly effectively residualize radiation in tumors means that
lower doses of radiation are needed to treat patients; thereby improving supply chain capability and cost of goods.
•Route of excretion from the body: Small molecules and peptides are primarily excreted in the urine rapidly passing
through from the blood into the bladder via the kidneys. Antibodies are cleared via the liver, which is a more radio-
tolerant organ.
In general, the properties of small molecules and peptides suit diagnostic targeted radiation agents, as the excess or
unbound radiation drug is rapidly lost from the body, resulting in a good contrast between the tumor and background
tissues and enabling favorable imaging within hours, allowing patients to be dosed and imaged within the same day.
Conversely, the high specificity of antibodies, along with their well validated, predictable characteristics in the body and
long retention in the tumor largely favor therapeutic use.
Radio Antibody-Drug Conjugate (rADC)
We refer to our antibody-based candidates as rADCs. These rADCs are radiopharmaceuticals that use an antibody as
both a homing device and a carrier to deliver a therapeutic radiation payload to a specific target. This property
differentiates them from chemotherapy, which cannot distinguish between healthy cells and tumor cells. rADCs are
designed to combine the targeting properties of monoclonal antibodies, which can discriminate between healthy and
cancerous tissue, with the cancer-killing capabilities of cytotoxic radiation.
We are pioneering a novel technology platform designed to optimize the therapeutic window for rADCs, which we refer
to as RADmAbs. This proprietary technology uses antibody engineering to modulate the pharmacokinetics of ‘full length’
antibodies such that they are designed to clear faster from the blood while maintaining the same high specificity to their
target and tumor localization properties. Since they retain the same overall structure as traditional antibodies, they also
share similar characteristics important for commercial development including a standard manufacturing pathway,
biological stability, immunogenicity and regulator familiarity.
In January 2025, we acquired patents, know-how and other intellectual property from ImaginAb comprising a proprietary
drug discovery platform, a pipeline of early-stage drug candidates against high-value targets, and several other novel
targets in discovery stage. The acquired intellectual property utilizes small engineered antibody formats that enable
highly specific cancer targeting, combined with fast tumor uptake and blood clearance.
We believe that this technology, alongside our other radiolabeling knowhow and technologies, can be applied to any
existing cancer-targeting antibody agent to potentially provide new intellectual property and a life-cycle management
option for prospective partners.
The Radiation Payload
The radioisotope is strongly bound to the target agent molecule either using traditional chemistry or trapping it using a
‘chemical cage’ called a 'linker' or 'chelator'. Different chelators are paired with certain isotopes, such as deferoxamine, a
linker that selectively binds with 89Zr (which we use in TLX250-Px), and the tetraxetan chelator, which binds isotopes like
177Lu (which we use in TLX591-Tx and TLX250-Tx) and 225Ac (which we use in TLX592-Tx and TLX252-Tx).
95
The choice of radioisotope and its decay profile impacts properties of the targeted radiation drug.
•Diagnostic radioisotopes for imaging: Radioisotopes emitting positrons can be detected by a PET camera. Gamma
emissions can be detected by a single photon emission computed tomography (SPECT) camera. These are
commonly referred to as “scanners.”
•Diagnostic radioisotopes for surgery: Both gamma and beta emitting radioisotopes can be used for the
intraoperative detection of tumors, using a handheld or robotic probe. The most commonly used isotope in radio-
guided surgery is 99mTc.
•Radioisotopes for therapy: Radioisotopes with the ability to kill cells for therapeutic effect are classified as either
beta- or alpha-emitters, based on their emission profile. Beta emitters (such as 177Lu and 131I) have a longer
penetration and may be more suitable for bulky metastatic disease. Alpha-emitters (such as 211At, 212Pb and 225Ac)
are substantially larger isotopes than beta-emitters and have the potential to deliver very high amounts of energy to
cancer cells in closer proximity to these particles, which can decrease the risk of damage to surrounding healthy
cells and increase the selectivity and potency of the radiation treatment. Alpha and beta therapies may be
complementary, with alpha therapies being more suitable for smaller or disseminated tumors (including micro
metastatic disease) and beta therapies being more suitable for treatment of bulkier tumors.
Our Prostate Cancer and PSMA programs
Overview
Our prostate cancer portfolio programs target PSMA, a protein that is overexpressed on the surface of prostate cancer
cells and is low or absent on most normal healthy cells. PSMA has become a major breakthrough in the staging,
treatment and management of prostate cancer. Imaging with targeted radiation can identify prostate cancer wherever it
is in the body and help guide patient treatment. The PSMA receptor is expressed in over 80% of prostate cancer tumors.
This expression of PSMA provides a specific target to design therapeutic and diagnostic agents for the treatment and
imaging of prostate cancer.
Market and Opportunity for Prostate Cancer Therapy
According to Pharma Intelligence, global incidence of prostate cancer was estimated to be 1,349,000 in 2022 and is
expected to reach approximately 1,455,000 by 2027 and in the U.S., the incidence of prostate cancer was estimated to
be 244,000 in 2022 and is expected to reach approximately 268,000 by 2027. The U.S. market opportunity for PSMA-
PET imaging agents in their approved indications is estimated to represent over US$2.5 billion per year, based on current
approved indications. The U.S. market opportunity for PSMA-targeted therapeutic agents is estimated at almost US$8
billion dollars per year based on Datamonitor patient-based forecasts for mCRPC and mHSPC.
High rates of screening in developed countries mean many men are diagnosed and treated early before their disease has
spread. These men receive local therapy, either prostatectomy or EBRT, and may be cured of their disease. However,
approximately 15% of patients develop advanced forms of the disease that can spread to other parts of the body. This is
known as metastatic prostate cancer.
According to Datamonitor's patient-based forecast, there were approximately 51,000 patients treated for mCRPC in the
U.S. in 2025. Approved treatment options for patients with mCRPC include androgen deprivation therapy, androgen
receptor pathway inhibitors, docetaxel chemotherapy, radium-223 for patients with bone-only metastases, PSMA-
targeted lutetium-therapy for patients having received prior docetaxel, and poly (ADP-ribose) polymerase (PARP)
inhibitors for patients with deleterious germline or mutated somatic homologous recombination repair gene. The global
market for systemic treatments for patients with mCRPC is estimated at approximately US$6.9 billion per year.
Pluvicto (lutetium (177Lu) vipivotide tetraxetan), marketed by Novartis, was approved by the FDA for the treatment of
patients with PSMA-positive mCRPC who have been treated with androgen receptor pathway inhibition and taxane-
based chemotherapy in March 2022. In March 2025, Pluvicto was approved by the FDA for earlier use prior to
chemotherapy, in PSMA-positive mCRPC. Pluvicto is the only FDA-approved PSMA-targeted therapy for the treatment of
prostate cancer. Novartis disclosed that Pluvicto recorded net sales of US$1,390 million in 2024 and US$1,994 million in
2025. Pluvicto uses a small-molecule approach to target the PSMA receptor and is administered in up to six cycles. In a
pivotal clinical trial, patients treated with Pluvicto showed an overall response rate of 30%, a median progression-free
survival of 8.7 months, and a median overall survival of 15.3 months.
Several other systemic radiotherapies are being investigated in clinical trials in the mCRPC setting and across other
stages of prostate cancer, and potentially could be commercialized in the future. We consider our most direct potential
competitors to be companies developing PSMA-targeted therapies in the mCRPC space, including Novartis, Convergent,
Point Biopharma, Eli Lilly & Company Ltd, Lantheus Holdings, Inc, Curium Holding France S.A.S, ARTBIO, Inc., Blue Earth
Therapeutics Ltd, Clarity Pharmaceuticals, Astra Zeneca PLC/Fusion Pharmaceuticals, Inc., Bayer AG, Orano Med SAS,
Isotopia Molecular Imaging Ltd, ITM Isotope Technologies Munich SE, Janssen Pharmaceuticals, AdvanCell Isotopes Pty
Ltd, Alpha-9 Theranostics, Cancer Targeted Technologies, FutureChem Co Ltd., Sinotau Pharmaceutical Group,
RadioPharm Theranostics, Precision Molecular, StarPharma, Ambrx Biopharm, Inc., Amgen Inc., Crescendo Therapeutics,
Poseida Therapeutics, Janux Therapeutics, Bivision Pharmaceuticals and Full-Life Technologies. Our competitors also
96
include companies developing other modalities to treat patients with mCRPC. (See “Business—Competition” for
additional information).
Market and Opportunity for Prostate Cancer Imaging
PSMA-PET imaging is used by clinicians to locate prostate cancer lesions and inform clinical decisions for patients.
PSMA-PET imaging is indicated in the U.S. for prostate cancer patients:
•with suspected metastasis who are candidates for initial definitive therapy;
•with suspected recurrence based on elevated serum PSA level; and
•for selection of patients who are indicated for PSMA-directed therapy as described in the prescribing information of
the therapeutic products.
We estimate that the PSMA-PET imaging market opportunity in the U.S. for these indications represents over 645,000
scans per year, which may be worth more than US$2.5 billion.
Guidelines and clinical research suggest potential future utilization of PSMA-PET imaging for:
•monitoring for progression in non-metastatic and mCRPC patients; and
•monitoring response to PSMA-directed therapy.
We estimate that these areas represent over 250,000 scans per year. We estimate that the combined addressable
market based on current and future guideline driven indications may be more than US$3.5 billion per year. Telix is
exploring 68Ga-PSMA-PET with Illuccix or Gozellix for diagnosis as a new indication, not currently FDA approved or
included in clinical guidelines. Combining 68Ga-PSMA-PET for diagnosis with current and guideline-driven indications, has
potential to bring the total addressable market to more than US$6.7 billion per year.
Our main competitors in the prostate cancer imaging market are companies with approved PSMA-PET diagnostics,
including Novartis, Lantheus Holdings, Inc. ("Lantheus"), Curium Pharma Ltd., Bracco Imaging S.p.A. (through its Blue
Earth Diagnostics affiliate) and Isotopia Molecular Imaging Ltd. Certain academic institutions, such as UCLA and UCSF,
also hold a license for a commercial PSMA-PET diagnostic.
In 2020, UCLA and UCSF obtained FDA approval for 68Ga-PSMA-11, which was the first PSMA-PET imaging agent to be
approved by the FDA. Pylarify (18F-piflufolastat), marketed by Lantheus, and Illuccix were subsequently approved by the
FDA in 2021. Locametz (68Ga-PSMA-11), marketed by Novartis, received FDA approval in 2022 and Posluma (18F-
flotufolastat), marketed by Blue Earth Diagnostics, received FDA approval in 2023. Gozellix was approved by the FDA in
March 2025. In August 2025, Lantheus announced that its NDA for a new formulation of its fluorine-18 based PSMA
imaging agent, had been accepted by the FDA with a PDUFA goal date of March 6, 2026. In May 2025, RadioMedix
submitted an Abbreviated New Drug Application ("ANDA") for 68Ga-PSMA-11. Several other PSMA-PET product
candidates are being evaluated in clinical trials for prostate cancer imaging and may be commercialized in the future.
Companies developing PSMA-PET imaging agents include ABX-CRO, Clarity Pharma Ltd., Curium Pharma Ltd., Isotopia
Molecular Imaging Ltd, Itelpharma, ITM Isotope Technologies Munich SE, Five Eleven Pharma, Fortis Therapeutics,
RadioMedix, HTA Co. Ltd and Jiangsu Hengrui Pharmaceuticals Co., Ltd.
Currently approved PSMA-PET imaging compounds use either a gallium-68 isotope (68Ga), such as Illuccix or Gozellix, or
a fluorine-18 isotope (18F) for PET imaging. Recent scientific publications illustrate evidence of important clinical
differences between 68Ga and 18F based imaging agents, including a lower rate of false positives with 68Ga, which can
potentially provide more accurate interpretation and understanding of the extent of disease (Fendler et al. EJNMMI.
2023; Kroenke et al. J Nucl Med. 2021). Also, 68Ga-based imaging agents have been shown to help clinicians detect
prostate cancer in patients with low disease burden (Burgard et al. Cancers. 2023). This early detection can lead to a
change in management and better outcomes for patients. Additionally, approved 68Ga-based imaging agents can use a
lower radiation dose than approved 18F-based agents, reducing radiation exposure for nuclear medicine physicians and
patients.
Therapy – TLX591-Tx
TLX591-Tx (lutetium (Lu177) rosopatamab tetraxetan) is a rADC directed at PSMA. We are evaluating the safety and
efficacy of TLX591-Tx in the ProstACT series of clinical trials. The key evidence from Phase 1 and Phase 2 studies
supporting the development of TLX591-Tx includes:
•Evidence that treatment with TLX591-Tx is well tolerated, including data from the Phase 1 ProstACT SELECT trial,
common grade 3 and 4 hematological events included thrombocytopenia, lymphopenia and neutropenia. All
hematological events were transient and manageable;
•Positive efficacy outcomes demonstrated following treatment of 242 patients across eight Phase 1 and Phase 2
clinical trials, including up to 42.3 months median survival in a single-arm Phase 2 clinical trial in 17 patients with
97
mCRPC when delivered under a fractionated dosing regimen. Data from Cohort 2 in the ProstACT SELECT trial
reported median rPFS of 8.8 months, an encouraging efficacy signal that should be considered directional in nature
given the small sample size in this Phase 1 clinical study;
•Evidence of low rates of off-target organ exposure observed in ProstACT SELECT; and
•Convenient two-dose regimen administered over 14 days with low radiation exposure.
As an rADC with an antibody targeting agent, we believe that TLX591-Tx may be differentiated from PSMA-targeted
therapies leveraging a small molecule approach as it has the potential for:
•Functional specificity for tumor-expressed PSMA, whereas small-molecule PSMA is taken up by endogenous PSMA;
•Reduced off-target radiation, with reduced potential for undesirable adverse events including dry eye, xerostomia,
and back pain from ganglia irradiation;
•Longer circulation time and tumor retention, while small molecule PSMA is rapidly excreted with approximately 70%
of activity lost after 12 hours; and
•Shorter dosing regimen of two doses, 14 days apart compared to dosing regimens lasting up to 30 weeks with
PSMA-targeted small peptide RLT.
Clinical Data
To date, 242 patients have been treated across eight Phase 1 and 2 trials of TLX591-Tx. We believe these data
cumulatively support the clinical validity of our intended fractionated dosing, which is designed to split a dose over a
longer treatment cycle to decrease toxicity without compromising efficacy. In an open-label, single-arm Phase 2 clinical
trial with six experimental dose cohorts of TLX591-Tx of 33 patients, we reported a 42.3 month median survival in 17
patients with advanced mCRPC treated at the higher dose level when TLX591-Tx was delivered under a fractionated
dosing regimen. Median survival was 19.6 months at the lower dose level and was 27.8 months across those dose
cohorts. At the higher dose level, 23.5% and 35.3% of patients had grade 3 and 4 neutropenia, respectively, and 29.4%
and 58.8% of patients had grade 3 and 4 thrombocytopenia, respectively. The trial met its primary endpoint, which was
to identify the maximum tolerated dose of TLX591-Tx when administered in two doses two weeks apart. The survival
benefits were a secondary endpoint. This trial did not contain a control group and was not powered to measure statistical
significance of the survival benefit, which is a limitation of single-arm trials.
ProstACT SELECT was a Phase 1 trial evaluating the safety and tolerability of TLX591-Tx in combination with standard of
care treatment in patients with mCRPC. The purpose of the ProstACT SELECT trial was to evaluate the ability of 68Ga-
PSMA-PET imaging to select patients for rADC-based PSMA therapy and to confirm the biodistribution of TLX591-Tx
when administered as two doses 14 days apart. The primary objectives of the study were to determine the whole body
distribution and organ radiation dose, and assess the safety and tolerability profile of TLX591-Tx, when administered in
combination with standard of care in second-line mCRPC. The evaluable population comprises a total of 30 patients
enrolled in the trial. In Cohort 1 comprising 5 patients, each patient received a 27 mCi dose followed by a 76 mCi
therapeutic dose for accuracy of biodistribution determination. In Cohort 2, 23 of 25 patients each received two
therapeutic 76 mCi doses. These patients included a heterogeneous patient population of low, medium and high disease
burden, with the majority of patients having undergone two prior lines of therapy.
Data from the ProstACT SELECT trial suggested evidence of high on-target PSMA tumor-binding and radiation delivery
to bone, nodal, and visceral metastases while minimizing uptake and toxicity in kidney and salivary glands. We believe
this biodistribution is significant when compared to small molecule therapeutic and diagnostic PSMA agents, which may
have greater off-target binding and subsequent radioactive dose to those organs.
We also observed evidence of consistent lesion delineation between TLX591-Tx and 68Ga-PSMA-11 imaging, within the
detection sensitivity and resolution limits of SPECT, evidence of uptake and retention in tumor and metastases up to 14
days post injection, the highest absorbed dose being in the liver (clearance organ) with minimal uptake in salivary glands,
and a long retention period that was evidence of internalization.
ProstACT SELECT Cohort 2 data also provides evidence of the potential clinical advantage of the short, simple treatment
regimen of two therapeutic doses administered 14 days apart. Hematologic treatment emergent adverse event ("TEAE")
rates were: Grade 3 thrombocytopenia (16% of patients, or 4/25), grade 3 neutropenia (28%, 7/25), grade 4
thrombocytopenia (20%, 5/25) and grade 4 neutropenia (4%, 1/25). All hematologic events were transient and
manageable. The trial demonstrated a median radiographic progression-free survival of 8.8 months, a secondary
objective, based on an evaluable patient population in Cohort 2 of 16 patients who each received two 76 mCi doses of
TLX591-Tx and did not have important protocol deviations. Based on these data, the trial appears to have achieved its
primary safety and tolerability objectives.
We are currently investigating TLX591-Tx in the Phase 3 ProstACT Global clinical trial in mCRPC patients previously
treated with one prior androgen receptor pathway inhibitor ("ARPI") in the metastatic castration-sensitive prostate
cancer ("mCSPC"), non-metastatic castration-resistant prostate cancer ("nmCRPC"), and first line (1L) mCRPC settings.
The trial is a multi-national, multi-center, prospective, randomized, controlled, open label study designed to investigate
98
and confirm the benefits and risks associated with TLX591-Tx a high-affinity PSMA-targeted rADC that delivers DNA
breaking radiation directly to PSMA-positive bone, nodal, or visceral metastases in patients with mCRPC. The trial will
enroll patients that have PSMA-positive mCRPC who have experienced disease progression following treatment with an
androgen receptor pathway inhibitor (abiraterone or enzalutamide) that was received in either the metastatic castration-
sensitive prostate cancer or first-line mCRPC treatment setting. The primary endpoint of the randomized portion of the
trial is radiographic progression-free survival and secondary endpoints include overall survival, objective response rate,
time to first symptomatic skeletal events, PFS, PSA decline of more than 50%, quality of life, and safety and tolerability.
In August 2025, this trial completed target enrollment of 30 patients in Part 1, a dosimetry and safety lead-in portion,
replicating the prior study using the product candidate specifications intended for commercial release. We dosed the first
patients in Part 2, the randomized treatment expansion arm, which we expect to enroll up to approximately 490 patients,
in Australia in December 2025. Part 1 data will be submitted to the FDA to obtain agreement to expand Part 2 of the trial
to U.S. sites. A public disclosure of preliminary results from Part 1 of the study will be aligned to engagement with the
FDA.
Therapy - TLX592-Tx (alpha-PSMA)
Through our TLX592 program, we are also exploring how the conjugation of an antibody vector with an alpha-emitting
isotope might deliver a next generation rADC with a different therapeutic profile. We believe that TLX592-Tx may be
suitable for patients with early-stage mCRPC with a low disease burden and for patients with late-stage mCRPC who are
no longer responding to lutetium therapy.
TLX592-Tx (64Cu/225Ac-RADmAb), is our next generation prostate cancer therapy candidate for targeted alpha therapy
and is our first clinical program based on our proprietary RADmAb-engineered antibody technology. The engineered
antibody vector is designed for faster elimination from circulation than standard antibodies. Therefore, it is expected to
reduce bone marrow residence time, mitigating the risk of hematologic toxicities while retaining PSMA-mediated tumor
localization and exertion of cytotoxic activity. TLX592-Tx is designed to be cleared by the liver without exocrine uptake.
We have conducted in vivo animal studies using an LNCaP (PSMA positive) tumor model and observed that treatment
with TLX592-Tx resulted in a significant improvement in survival time of nude mice compared to a phosphate buffered
saline treated control group (Fletcher et al. Mol Pharmaceutics. 2025). We studied the toxicological profile in CD1 mice
and did not observe any treatment-related toxicity up to the highest dose level (single administration).
Clinical Data
Prior to commencing therapeutic studies with the alpha emitting isotope 225Ac, we conducted the Phase 1 CUPID trial in
which we evaluated TLX592-Tx radiolabelled with the imaging isotope 64Cu, in patients with advanced prostate cancer.
We used 64Cu to understand the safety profile, pharmacology and dosimetry prior to the use of 225Ac, since 64Cu is
detectable by PET, whereas 225Ac is not. We investigated patients with PSMA avid disease based on Illuccix imaging,
across three mass dose levels, to assess the safety profile, pharmacokinetics, biodistribution and dosimetry. Based on
data from 11 evaluable patients, we observed accelerated elimination from blood circulation compared to the standard
antibody used in TLX591-Tx, with similar on-target and off-target biodistribution. There were no treatment-related
adverse events observed in the trial.
In July 2025, we received regulatory approval in Australia to commence the Phase 1 FIH therapeutic trial, AlphaPRO,
designed to evaluate the safety profile of 225Ac-labelled TLX592-Tx.
Therapy - TLX597-Tx (small molecule-based targeted radionuclide therapy)
TLX597-Tx (177Lu-DOTA-HYNIC-panPSMA) is our novel small-molecule lutetium-PSMA therapy being developed to
enable access for prostate cancer patients in select geographies.
Clinical Data
Initial data from an Australian IIT showed TLX597-Tx could be effective with reduced salivary gland uptake versus
current standard of care, and a retrospective translational study of 145 patients highlighted positive efficacy outcomes
versus a control group (Luna-Gutiérrez et al. Pharmaceutics. 2023). In the IIT, early dosimetry data from 12 of 120
patients suggests TLX597-Tx delivers approximately one-third of the radiation dose to the salivary glands and one-half
to the kidney, compared to the current approved PSMA-directed lutetium therapy. We believe this could lead to reduced
incidence of acute renal injury and xerostomia (dry mouth), while facilitating dose intensification versus current standard
of care, to improve efficacy.
This candidate is being developed to enable access to lutetium therapy in select geographies, and is the subject of the
ongoing Phase 2 OPTIMAL PSMA IIT, which is expected to enroll 120 mCRPC patients in Australia.
Imaging – Illuccix
Illuccix (also referred to as TLX591-Px in territories where approval has not yet been granted, 68Ga-PSMA-11) is a
preparation for imaging prostate cancer with PET.
99
The “cold kit” format of Illuccix enables rapid radiolabeling at room temperature with high radiochemical purity and
production consistency, suited to the commercial and hospital radiopharmacy setting. Illuccix is approved in the U.S.,
Australia, Austria, Belgium, Brazil, Canada, Cyprus, Czech Republic, Denmark, Finland, France, Germany, Greece, Ireland,
Italy, Luxembourg, Malta, Netherlands, Norway, Portugal, Slovakia, Spain, Sweden and the United Kingdom. Approved
indications for patients with prostate cancer include staging of high-risk patients, identification of suspected recurrence,
and selection for PSMA-directed therapy. We are also exploring potential future utilization in additional indications for
prostate cancer patients through our lifecycle management program. These include monitoring progression in metastatic
and non-metastatic castration resistant patients, monitoring response to PSMA-directed therapy, and as an adjunct to
MRI for the diagnosis and detection of prostate cancer.
The key evidence supporting the use of Illuccix include:
•Broad availability in the U.S. through over 225 radiopharmacies and with flexible scheduling;
•Validated accuracy compared to other PSMA imaging agents, including lower rate of false positives and efficacy in
patients with low disease burden; and
•Potential for expanded clinical utility based on guidelines and clinical research.
In May 2025, our pivotal Phase 3 registration study of TLX591-Px for prostate cancer imaging in Chinese patients
completed target patient enrollment. Illuccix China is a prospective, open-label, single-arm, multicenter study conducted
in collaboration with our strategic partner for the Greater China region, Grand Pharma. The study, which enrolled 140
patients, aims to demonstrate equivalence of TLX591-Px in imaging prostate cancer in Chinese and Western patients, in
order to bridge to the marketing authorization granted to Illuccix by the FDA. In December 2025, Telix and Grand Pharma
announced preliminary data from the Illuccix China study, indicating that the primary endpoint had been met. The data
was used to file a new drug application for TLX591-Px with the Chinese National Medical Products Administration
("NMPA") Center for Drug Evaluation ("CDE"), which was accepted for review in January 2026.
In January 2026, we announced that the first patient had been dosed in our pivotal Phase 3 registration study of TLX591-
Px for prostate cancer PET imaging in Japanese patients. Illuccix Japan is a prospective, open-label, single-arm, intra-
patient, multicenter study designed to evaluate the visualization efficacy and safety profile of 68Ga-PSMA-11 PET/CT
compared with conventional imaging (computed tomography ("CT") and bone scintigraphy) in Japanese patients with
biochemically recurrent ("BCR") prostate cancer following prior radical prostatectomy. The study will enroll up to 105
Japanese men at up to 10 sites and data is intended to support a future marketing authorization application for TLX591-
Px in Japan.
Imaging – Gozellix
Gozellix (68Ga PSMA-11) is Telix's next generation cold kit for the preparation of PSMA-PET imaging for prostate cancer,
and was approved by the FDA in March 2025. Gozellix is designed to have an extended distribution profile compared to
currently approved 68Ga PSMA-PET imaging agents due to the use of 68Ga sourced from newer high activity generators
and cyclotrons.
We believe that Gozellix may further expand the availability and distribution of PSMA-PET imaging due to its longer shelf
life and resulting expanded distribution radius. We believe that Gozellix has the potential to address unmet needs by
extending availability of PSMA-PET imaging to substantially all PET/CT locations in the U.S. Many PET/CT imaging sites
that are not served by approved PSMA-PET imaging agents are located in rural and underserved areas.
Gozellix was granted transitional pass-through payment status by CMS, effective October 2025 for a three-year period.
This status enables CMS to provide separate payments for Gozellix and the PET-CT scan when performed in the hospital
outpatient setting.
We are also exploring potential future utilization in additional indications for prostate cancer patients through our
lifecycle management program. These include monitoring progression in metastatic and non-metastatic castration
resistant patients, monitoring response to PSMA-directed therapy and as an adjunct to MRI for the diagnosis and
detection of prostate cancer.
Imaging – TLX593-Px
In June 2025 we launched a novel PET radiochemistry solution based on 18F-aluminum fluoride ("AlF"), named AlFluor™.
As part of AlFluor’s development, we signed a strategic agreement with University Hospital Ghent and Ghent University
for a novel [18F]AlF-PSMA-11 targeting agent, which we have designated as TLX593-Px. The agreement includes a
comprehensive chemistry, manufacturing and controls ("CMC") package suitable for the preparation of a Drug Master
File ("DMF") and provides exclusive access to TLX593-Px clinical safety and efficacy data, including a Phase 3 trial in 96
prostate cancer patients, where PSMA-11 (gozetotide) was labeled interchangeably with 18F and 68Ga.
100
Bone Metastases and Pain Palliation
TLX090-Tx
TLX090-Tx (153Sm-DOTMP) is a novel kit-based bone-seeking targeted radiopharmaceutical product candidate that uses
a next generation chelating agent to deliver a proprietary formulation of Samarium-153 radioisotope. It is a combination
of patented, lower specific activity form of Samarium-153, a beta-emitting radioisotope with a 46-hour half-life, and the
chelating agent DOTMP (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetramethylene-phosphonate), which selectively
targets sites of high bone mineral turnover, a known characteristic of bone metastases, and minimizes off-target
migration.
We believe that TLX090-Tx may be administered as a single dose, multiple doses and higher dose regimens for pain
management of bone metastases and osteosarcoma therapy, including in pediatric patients. We believe that TLX090-Tx
is highly aligned with our existing therapeutic focus areas of prostate cancer, glioma and sarcoma.
Clinical Data
In August 2021, the FDA cleared the IND application to commence Phase 1, open-label, dose escalating study for
TLX090-Tx (originally CycloSam®) in patients with osteosarcoma and other solid tumors metastatic to bone, with the
primary objective of establishing the maximum tolerated dose ("MTD") and evaluating pharmacokinetics, dosimetry, and
safety. Patients received an imaging dose of 0.5 mCi/kg on Day 1 and then a therapeutic dose on Day 8, with escalation
planned up to 3.0 mCi/kg. A total of five patients were enrolled and treated in the first two cohorts (three patients at 0.5
mCi and two patients at 1 mCi/kg). SPECT/CT scans of these patients showed that TLX090-Tx was highly targeted to
bone and had preferential uptake in bone tumors, with no evidence of soft tissue activity and rapid elimination via the
kidney and bladder. Complete blood count and comprehensive metabolic panel blood testing data indicated no toxicities
or drug-related adverse events, some mild and transient drop in white blood cell counts that recovered by Week 6
without intervention, and there were no clinically significant changes in liver and kidney function. No transfusions or stem
cell recovery procedures were necessary. While pain relief was observed in patients, this was exploratory and not a
primary endpoint. Visual analogue scale pain scores taken at baseline and then weekly after dosing suggest fast-acting,
long-lasting pain relief, improved mobility and improved quality of life. We believe that pain relief is evidence of that
TLX090-Tx may not have similar risks or the potential adverse events of opiate pain medications, and may offer a viable
alternative for pain palliation in patients with advanced cancer and painful osteoblastic bone metastases.
We believe that TLX090-Tx has the potential to deliver significant improvements to existing bone-seeking agents for the
palliation of painful osteoblastic bone metastases in late-stage metastatic disease. We believe TLX090-Tx may enable
the pain management of prostate cancer bone metastases, where there remains a significant unmet medical need
particularly after progression from other forms of radionuclide and radiation therapy. We also believe that TLX090-Tx
may benefit patients with metastatic lung and breast cancer, where many patients develop painful bone metastases, and
disease management often focuses on quality-of-life palliative care.
TLX090-Tx has also been granted orphan drug and rare pediatric disease designations by the FDA for the treatment of
osteosarcoma. These designations apply only to the osteosarcoma indication and do not apply to the current pain
palliation indication being pursued in the SOLACE trial. The rare pediatric disease designation may enable TLX090-Tx to
be brought to market more rapidly for osteosarcoma through regulatory incentives, including eligibility for a pediatric rare
disease priority review voucher that may be applied to this or other programs. The orphan drug designation and the rare
pediatric disease designation do not increase the likelihood of marketing approval.
In October 2025, we dosed the first patient in an open-label Phase 1 clinical trial designed to evaluate the
pharmacokinetics, dosimetry, safety, and pain palliation of TLX090-Tx. SOLACE (Samarium Optimized for Long-lasting
Analgesia in Cancerous End-stage bone pain) is expected to enroll up to 33 patients with advanced cancer and painful
osteoblastic bone metastases. Data from SOLACE aims to establish clinical comparability to legacy 153Sm treatments,
which in turn is expected to support a streamlined registration pathway as an analgesic, paving the way for a much-
needed, non-opioid solution for patients living with bone pain in the late stages of advanced cancer.
Our Kidney Cancer and CAIX programs
Overview
CAIX is a protein expressed on the surface of ccRCC and other solid tumors, including bladder or urothelial, breast, brain,
cervix, colon, esophagus, head and neck, lung, ovarian, pancreatic and vulval cancers. CAIX is overexpressed in over
95% of ccRCC tumor cells and has limited expression on healthy tissue.
CAIX is often expressed in hypoxic (oxygen deficient) tumor cells. Hypoxic tumors are also typically more aggressive and
less responsive to current treatments, particularly immunotherapies. A published study has shown that tumor sections
from patients that failed to respond to PD-1 blockade therapy showed significantly higher CAIX expression than those
that responded (n = 19), suggesting that CAIX expression is associated with poor response to immunotherapy.
Furthermore, a published study has demonstrated that in 117 hepatocellular carcinoma patients, positive CAIX expression
correlated with reduced disease-free survival and overall survival.
101
We believe the association between hypoxia, disease progression, and therapeutic resistance underscores the relevance
of this target. Whereas normal endogenous expression of CAIX is very low, CAIX has been found to be differentially
expressed on Tregs in the tumor micro-environment in a number of solid tumors. We are developing products for the
detection and treatment of ccRCC and investigating the potential of CAIX as a pan-cancer target in multiple tumor types.
Market and Opportunity for Kidney Cancer Therapy
We estimate that over 25% of ccRCC patients, equivalent to over 16,000 patients per year in the U.S., have metastatic
RCC. Approved treatment options for ccRCC patients include immunotherapy, tyrosine kinase inhibitors, mTOR inhibitors,
and HIF-2α inhibitors. The U.S. market for systemic RCC treatment is estimated to be approximately US$6.7 billion per
year based on Datamonitor’s patient-based forecast, and the global market is estimated at approximately US$9.2 billion.
We are exploring the use of TLX250-Tx for the treatment of ccRCC, either in combination with an immunotherapy or as a
monotherapy, to treat metastatic disease expressing the CAIX receptor. There is a significant need for new therapeutic
options for patients with advanced kidney cancer, given its inherent resistance to conventional chemotherapy. Despite
the transformative impact of immunotherapies on the prognosis of patients with metastatic kidney cancer, a considerable
number fail to respond adequately and eventually progress.
An increasing body of scientific evidence suggests low doses of targeted radiation can potentially overcome immune
resistance. This approach, known as immunological “priming,” has the potential to render tumors more susceptible to
cancer immunotherapy. Several pre-clinical studies have shown an enhanced therapeutic outcome of checkpoint
inhibitors when they are administered after a systemic radiotherapy, including rendering immunologically inert tumors
sensitive to treatment.
There is currently no CAIX-targeted radiopharmaceutical therapy approved to treat ccRCC. Several other systemic
radiotherapies are being investigated to treat ccRCC targeting CAIX, and potentially could be commercialized in the
future.
We consider our most direct competitors to be companies developing CAIX-targeted systemic radiotherapies, including
ITM Isotope Technologies Munich SE/Debiopharm SA, Precision Molecular, Inc., Bristol Myers Squibb Company/RayzeBio,
Inc., AstraZeneca PLC/Fusion Pharmaceuticals Inc. and PeptiDream Inc. Our competitors will also include companies
developing other modalities to treat ccRCC.
Market and Opportunity for Kidney Cancer Imaging
According to the Global Cancer Statistics 2022: GLOBOCAN survey, global incidence of kidney cancer was 434,840 in
2022. In the U.S., the incidence of kidney cancer was 81,800 in 2022 according to the American Cancer Society.
Approximately 80-90% of malignant kidney tumors are ccRCC. It is one of the subtypes with the worst prognosis and
survival often depends on how early it is detected.
Kidney cancer is typically discovered incidentally and diagnosed using a number of modalities including CT scanning, MRI
scanning, ultrasound, and biopsy.
The detection of renal masses is increasing due to widespread use of cross-sectional imaging. Many of these are small
and represent a diagnostic challenge as current imaging techniques, including ultrasound and MRI, cannot reliably
distinguish benign or malignant lesions from renal cell carcinoma, leading to invasive biopsy or partial nephrectomy
(kidney removal) to confirm the diagnosis. These procedures are cumbersome and often lead to complications.
Currently, there are major unmet needs for the improvement in diagnosis of ccRCC from indeterminate renal masses as
well as improving the staging of more advanced ccRCC through more accurate and specific imaging techniques. In the
U.S., we estimate that there are at least 116,000 patients per year with renal masses that could require a biopsy or
nephrectomy. We believe that an additional 50,000 patients with ccRCC could benefit from more accurate staging or
improved identification of recurrence using molecular imaging. This market is estimated to represent approximately
US$1.5 billion per year. We also believe that there may be patients that may benefit from more than one scan and from
active surveillance.
Currently, there is no approved agent for CAIX imaging. We consider our most direct competitors to be companies
developing ccRCC or CAIX-targeted imaging agents, including ITM Isotope Technologies Munich SE/Debiopharm SA,
Philogen S.p.A., Precision Molecular, Inc., PeptiDream Inc., Norroy Bioscience Co. Ltd., and Five Eleven Pharma. Our
competitors will also include companies developing other modalities to image ccRCC and CAIX.
Therapy – TLX250-Tx
TLX250-Tx (177Lu-DOTA-girentuximab) is a rADC therapeutic product candidate for the treatment of kidney cancer.
TLX250-Tx is currently under investigation for the treatment of patients with ccRCC in an investigator-initiated trial in
combination with checkpoint and tyrosine kinase inhibitors in up to 100 patients (Phase 1b/2 STARLITE-1). In October
2025 we received ethics approval in Australia to commence LUTEON, a global Phase 2/3 trial of TLX250-Tx as a
monotherapy in participants with recurrent ccRCC, who have progressive disease on or after two and no more than three
prior lines of therapy.
102
We are using girentuximab to target CAIX as it is designed to have a high degree of selectivity and affinity for the target
and is cleared from the body by the liver. The lack of kidney excretion is an advantage for patients with primary kidney
disease. We believe the target profile and the properties of girentuximab make the ccRCC phenotype promising as the
first therapeutic indication for TLX250-Tx. We believe the therapeutic potential of TLX250-Tx may also extend into other
cancers that significantly express CAIX, including certain VHL-induced cancers, ovarian cancer, triple-negative breast
cancer and bladder cancer. We believe that our preliminary clinical data in triple-negative breast cancer and bladder
cancer supports future development of TLX250-Tx in these indications.
Clinical Data
TLX250-Tx is being evaluated for the treatment of patients with ccRCC, both as a monotherapy and in combination with
checkpoint inhibitors.
In October 2025 we received ethics approval in Australia to commence LUTEON, a global Phase 2/3 trial of TLX250-Tx
as a monotherapy in advanced metastatic ccRCC. This global, multicenter, open-label trial will be conducted in two parts,
a safety and dosimetry lead-in (Part 1), followed by randomized efficacy assessment (Part 2). The objective of Part 1 is to
determine the recommended Phase 3 dose ("RP3D") for use in Part 2.
STARLITE-1 is a single arm Phase 1/2 IIT of TLX250-Tx in combination with cabozantinib and nivolumab in treatment
naïve patients with advanced ccRCC. The trial, which has a target enrollment of 100 patients, is sponsored by the MD
Anderson Cancer Center and commenced dosing patients in September 2025.
We believe that tumor-targeted radiation stimulates remodeling of the tumor micro-environment and can kill
immunosuppressive cells and stimulate T-cell recruitment to attack tumor cells. This immune re-programming may
increase the therapeutic response to treatment with checkpoint inhibitors.
TLX250-Tx was previously the subject of the exploratory Phase 2 STARLITE-2 IIT in combination with nivolumab, and the
Phase 1b STARSTRUCK trial in combination with peposertib, a DNA-dependent protein kinase ("DNA-PK") inhibitor in
collaboration with Merck KGaA, Darmstadt, Germany. We are now initiating the LUTEON study, a global Phase 2/3 trial of
TLX250-Tx as a monotherapy in treatment‑refractory disease. We believe LUTEON will generate the pivotal data
required to define the optimal therapeutic activity, schedule, and treatment window for TLX250-Tx. These data are
essential to inform any future development including combination regimens. In light of this, we have made the decision to
close STARLITE-2 and STARSTRUCK trials at this time to ensure that any future combination‑therapy development is
aligned with the most robust and clinically validated dosing framework.
Therapy – TLX252-Tx
In our TLX252-Tx (225Ac-DOTA-girentuximab) program, we are developing girentuximab radiolabeled with the alpha-
emitting isotope actinium-225 as a potential follow-on for the TLX250-Tx (beta) program by addressing unmet need in
radiation-resistant CAIX-positive disease. TLX252-Tx has demonstrated pre-clinical proof-of-concept in several
published studies (Merkx et al. Pharmaceuticals. 2022), which we believe supports the initiation of an administered-
activity escalation trial of TLX252-Tx for patients with advanced metastatic kidney cancer and other CAIX-expressing
tumors.
We expect that data from our existing CAIX-targeting programs (TLX250-Px diagnostic and TLX250-Tx therapy) will
complement and inform the clinical and regulatory development strategy for TLX252-Tx. In September 2025 we received
ethics approval in Australia to commence ALPHIX, a first-in-human Phase 1 study of TLX252-Tx in patients with
advanced metastatic kidney cancer and other CAIX-expressing tumors. CAIX is a broad indicator of aggressive,
treatment-resistant disease in most solid tumors and we believe that the use of an alpha-emitter like 225Ac for CAIX-
targeted radiation may help overcome treatment resistance in these aggressive tumors given the unique properties of α-
particles, which make this treatment modality impervious to conventional cellular resistance mechanisms.
Imaging – TLX250-Px
TLX250-Px (89Zr-DFO-girentuximab) is a PET imaging candidate for the characterization of renal masses as ccRCC. We
evaluated TLX250-Px in the Phase 3 ZIRCON trial in 300 patients, of which 284 were evaluable. The trial met all primary
and secondary endpoints, including showing 86% sensitivity and 87% specificity and a mean positive predictive value of
93% for ccRCC across three independent readers. We believe this demonstrated the ability of TLX250-Px to reliably
detect the clear cell phenotype and provide an accurate, non-invasive method for diagnosing ccRCC. TLX250-Px was
granted breakthrough therapy designation by the FDA in 2020.
We submitted a BLA for TLX250-Px to the FDA for regulatory approval in December 2023. The BLA was granted on a
rolling review process. We completed the BLA submission in May 2024, and in July 2024, the FDA declined to approve
the BLA and issued an RTF determination. The denial was based on a filing concern related to demonstrating adequate
sterility assurance during dispensing of TLX250-Px in the radiopharmacy production environment. In December 2024 we
resubmitted our BLA to the FDA for TLX250-Px, and the FDA accepted our application in February 2025. In August 2025,
the FDA issued a CRL, citing deficiencies relating to the CMC package. The FDA requested additional data to establish
comparability between the drug product used in the ZIRCON Phase 3 clinical trial and the scaled-up manufacturing
process intended for commercial use. Additionally, the FDA documented notices of deficiency (Form 483) issued to two
third-party manufacturing and supply chain partners that required remediation prior to resubmission. In December 2025
103
we participated in a Type A meeting with the FDA to align on plans to address CMC deficiencies cited, and in January
2026 we participated in an additional Type A meeting to align on plans to address comparability between the clinical
drug product and that intended for commercial use. Following these meetings, Telix believes it has alignment with the
Agency on all key resubmission aspects, and is working both internally and with third-party vendors to implement these
items for resubmission. While we believe that the planned remediation of the BLA for TLX250-Px will meet FDA
requirements, there can be no assurance that, once resubmitted, the FDA will accept our BLA for review, or approve
TLX250-Px. If approved, TLX250-Px would be the first targeted radiopharmaceutical imaging agent for kidney cancer to
be approved in the U.S.
The key attributes supporting development of TLX250-Px include:
•High affinity for CAIX, expressed in up to 95% of ccRCC and many hypoxic solid tumors, low expression in normal
tissue;
•Positive results in Phase 3 ZIRCON trial including key secondary endpoints that demonstrated detection of ccRCC
even in small renal masses (less than 4cm); and
•Breakthrough therapy designation from the FDA granted in 2020.
Breakthrough therapy designation may not lead to a faster development or regulatory review or approval process, and
does not increase the likelihood that TLX250-Px will receive marketing approval.
Clinical Data
In 2022, we completed the pivotal Phase 3 ZIRCON trial evaluating TLX250-Px in 300 patients, and primary results were
published in Lancet Oncology in 2024 (Shuch et al. Lancet Oncol. 2024). The trial met all primary and secondary
endpoints, including showing 86% sensitivity and 87% specificity and a 93% PPV for ccRCC across three independent
readers. We believe this trial demonstrated the ability of TLX250-Px to reliably detect the clear cell phenotype and
provide an accurate, non-invasive method for diagnosing ccRCC. Confidence intervals exceeded expectations in all three
readers, showing evidence of high accuracy and consistency of interpretation.
The data from the trial demonstrated the ability of TLX250-Px to characterize renal masses as ccRCC, which could
support improved clinical decision making and limiting the need for invasive procedures like biopsies and nephrectomies.
A total of 300 patients were dosed with TLX250-Px in the trial and 284 patients had a central histology reading and
evaluable TLX250-Px PET scan at central review.
The study also met the key secondary endpoint, achieving 85% sensitivity and 89% specificity in detecting ccRCC in
tumors ≤4cm (T1a classification), currently a significant clinical challenge in the diagnosis of ccRCC. In very small renal
lesions (≤2cm, a secondary endpoint), sensitivity was 84% for all three independent readers, with specificity ranging
from 90.0% to 100%.
The table below provides a breakdown of the three independent reader scores, overall score and confidence intervals of
the full analysis set.
Reader 1 | Reader 2 | Reader 3 | Overall % (95 % CI) | |
Sensitivity, % | 84.13 | 85.19 | 87.30 | 85.50 |
Lowest bounds, Wilson 95% CI | 78.24 | 79.42 | 81.80 | (79.80; 89.80) |
Specificity, % | 88.42 | 88.42 | 84.21 | 87.00 |
Lowest bounds, Wilson 95% CI | 80.45 | 80.45 | 75.57 | (78.80; 92.30) |
Positive predictive value, % | 93.53 | 93.60 | 91.67 | 93.00 (88.00; 96.00) |
Negative predictive value, % | 73.68 | 75.00 | 76.92 | 75.00 (66.00; 82.00) |
Accuracy, % | 85.56 | 86.27 | 86.27 | 86.00 (81.50; 89.60) |
The majority of adverse events in the trial were post-surgical complications and not treatment related. A total of 261
treatment-emergent adverse events were reported in 122 of 300 patients (40.7%), of which 146 were mild, 50 were
moderate and 49 were severe. Four of the treatment-emergent adverse events were life-threatening and one was fatal.
13 treatment-emergent adverse events were considered to be treatment related, of which, nine occurred before surgery
and four occurred after surgery. No unexpected safety signals were observed and tolerability data were consistent with
experience of girentuximab in previous therapeutic and imaging studies.
In November 2024, we dosed the first patient in the Phase 3 ZIRCON-CP study of TLX250-Px PET imaging of ccRCC in
Chinese patients. The study, which will enroll up to 82 patients, is being conducted in collaboration with the Company’s
strategic partner for the Greater China region, Grand Pharmaceutical Group Limited ("Grand Pharma"), to demonstrate
104
that the diagnostic utility of TLX250-Px is equivalent in Chinese and Western populations. The clinical data from ZIRCON-
CP is intended to support future marketing authorization applications for this breakthrough technology in the region.
In October 2024, we announced that we dosed the first patient in the Phase 2 CA-NINE trial, which is an investigator-
initiated Phase 2 trial evaluating TLX250-Px in patients with ccRCC after surgery. The trial plans to enroll 91 patients with
intermediate-to-high risk ccRCC post-surgery and is designed to identify ccRCC where it has recurred, including
metastatic disease, and may inform future label expansion for TLX250-Px.
There are several other investigator-led trials of TLX250-Px that have recently completed enrollment, including the
Phase 1 ZiP-UP trial in patients with metastatic urothelial carcinoma or bladder cancer, the Phase 2 OPALESCENCE trial
in patients with triple-negative breast cancer, and the Phase 1 PERTINENCE trial in patients with non-muscle invasive
bladder cancer ("NMIBC"). The OPALESCENCE and PERTINENCE trials reported positive preliminary data during 2022 at
the European Association of Nuclear Medicine Annual Congress, with early results suggesting theranostic potential in
these difficult to treat diseases. In May 2025, final positive results from the PERTINENCE trial along with preclinical data
were published in Cancers demonstrating the promising therapeutic role of 211At-girentuximab in NMIBC. In November
2025, final positive results from the OPALESCENCE trial were published in the European Journal of Nuclear Medicine and
Molecular Imaging, demonstrating the potential for TLX250-Px to detect metastatic triple negative breast cancer
("TNBC") lesions that may be resistant to chemotherapy and therefore have a more aggressive profile resulting from
hypoxia.
In November 2025, we announced results from the ZIRCON-X study which found that almost half of all patients imaged
with TLX250-Px would have undergone a change in clinical management, when compared with baseline SOC imaging.
ZIRCON-X was a non-interventional, prospective, post-hoc study sponsored by Telix – using imaging data from Telix’s
parent pivotal Phase 3 ZIRCON study – that assessed the impact of TLX250-Px imaging on clinical decision-making
versus SOC contrast-enhanced diagnostic imaging in 294 patients with indeterminate renal masses ("IRMs"). The study
found that 143 patients (48.6%) would have undergone a change in clinical management if imaged with TLX250-Px, and
over 20% of these patients (31 out of 143) could have potentially avoided invasive biopsy. Of all evaluable patients, more
than one third (110 out of 294, or 37.4%) would have had a major change in clinical management based on defined
categories, with approximately 30% having their treatment escalated or de-escalated. A subset of 18 patients initially
selected for active surveillance would have been escalated to immediate treatment.
Our Brain Cancer Programs and LAT1/LAT2
Overview
According to the Global Cancer Statistics 2022: GLOBOCAN survey, global incidence of brain and nervous system tumors
was 321,731 in 2022. Gliomas make up approximately 30% of all brain and central nervous system tumors and 80% of all
malignant brain tumors. In the U.S., according to the 2025 CBTRUS Statistical Report, the incidence of malignant glioma
was 22,836.
Glioblastoma is the most aggressive sub-type of glioma, representing 14,190 cases per year in the U.S. It has a poor
prognosis, primarily due to there being few effective treatment options. Glioblastoma has a median survival from initial
diagnosis of 12-15 months with a 5-year survival of 7%.
The mainstay of treatment for glioblastoma is surgical resection, followed by combined radiotherapy and chemotherapy.
Despite such treatment, recurrence occurs in almost all patients. Our brain cancer program targets membrane transport
proteins called LAT1 and LAT2, which are important targets in cancer development as they supply tumors with essential
amino acids, promoting cell proliferation, angiogenesis and mediating drug and nutrient delivery across the blood-brain
barrier. LAT1 and LAT2 are highly expressed in the blood-brain barrier and in various types of cancer, including
glioblastoma.
Market and Opportunity for Brain Cancer Treatment
While surgical resection plus radiation therapy are the mainstays of treatment, the vast majority of patients experience
disease recurrence. Thus, there remains an important need for therapies targeted towards glioblastoma in patients in
both the front-line treatment setting, as well as for patients experiencing disease recurrence following surgical
intervention.
There are several systemic radiotherapies being evaluated in clinical trials for the treatment of glioblastoma. We consider
our most direct competitors to be companies developing systemic radiotherapies for brain tumors, including ITM Isotope
Technologies Munich SE, Molecular Targeting Technologies, Inc., EvaThera Theranostics, Novartis, RadioPharm
Theranostics, Plus Therapeutics and Cellectar Biosciences, Inc. Our competitors will also include companies developing
other modalities to treat brain cancer.
Market and Opportunity for Brain Cancer Imaging
We believe there are a number of opportunities to address unmet needs in the market for imaging of glioma. The first is
improving the characterization of recurrence. Although MRI is the current standard of care for imaging of glioma patients,
the accurate identification of recurrence remains an important unmet medical need. The U.S. market opportunity for
imaging in this setting is estimated at 20,000 scans per year. This market is estimated to represent approximately
US$100 million to US$140 million per year.
105
The second is improving adjuvant radiation treatment planning in glioblastoma patients, which is also an important unmet
medical need. The U.S. market opportunity imaging in this setting is estimated to be 15,000 scans per year.
The third opportunity is improved identification of recurrence in patients with brain metastases. The incidence of brain
metastases in the U.S. is estimated to be approximately 168,000 cases per year. The U.S. market opportunity for imaging
in this setting is estimated at approximately 100,000 scans per year. This aggregate market is estimated to represent
approximately US$600 million per year.
There are several molecular imaging agents being evaluated in clinical trials for the imaging of glioma and brain
metastases. We consider our most direct competitors to be companies developing imaging agents for brain tumors,
including Novartis, Blue Earth Diagnostics, RadioPharm Theranostics, Curasight, Molecular Targeting Technologies, Inc.,
and EvaThera Theranostics. Our competitors could also include companies developing other modalities to image brain
cancer.
Therapy – TLX101-Tx
TLX101-Tx (iodofalan 131I) is our lead therapeutic product candidate for the treatment of patients with brain cancer that
targets the LAT1 receptor. TLX101-Tx is a novel approach that is designed to readily pass through the blood-brain
barrier, the normal protective barrier that prevents many potential drug candidates from entering the brain.
We are currently evaluating TLX101-Tx in front line and recurrent glioblastoma in the IPAX series of trials. TLX101-Tx has
been granted orphan drug designation in the U.S. and Europe for the treatment of glioma. Orphan drug designation may
not lead to a faster development or regulatory review or approval process and does not increase the likelihood that
TLX101-Tx will receive marketing approval.
The key attributes supporting development of TLX101-Tx include:
•IPAX-1 Phase 1 trial, demonstrated evidence of tumor responses in recurrent glioblastoma including some patients
with prolonged disease stabilization, and evidence of rapid clearance of TLX101-Tx from the brain;
•IPAX-2 Phase 1 trial extended TLX101-Tx into the front-line setting, building upon experience in recurrent setting;
•TLX101-Tx has been granted orphan drug designation in the U.S. and Europe for the treatment of glioma.
In 2025, we received regulatory approval in Australia and the EU to commence the Phase 3 IPAX-BrIGHT pivotal trial of
TLX101-Tx with sites open for enrollment. IPAX BrIGHT is designed to assess the safety and tolerability of TLX101-Tx in
combination with chemotherapy (lomustine), compared to chemotherapy alone. The global, multicenter, open-label study
is enrolling patients with radiographically confirmed recurrent glioblastoma at first recurrence. The trial consists of two
parts: Part 1, a safety and dosimetry lead-in with a minimum of 18 patients, and Part 2, randomized efficacy assessment,
with enrollment numbers to be determined following Part 1. Patient dosing in IPAX-BrIGHT will mark the first targeted
radionuclide therapy to enter Phase 3 development for recurrent glioblastoma.
Clinical Data
In 2022, we reported the final results from the IPAX-1 Phase 1/2 trial evaluating TLX101-Tx in combination with EBRT in
patients with recurrent glioblastoma. In 2024, results were published in Neuro-Oncology Advances, confirming the trial
met its primary safety and tolerability objective.
We enrolled ten patients in the trial, nine of whom received the full dose of ~2GBq (2000 MBq) of TLX101-Tx, either in
the form of a single administration or one of two triple-fractionated regimens. All dosing regimens were well tolerated.
Dosimetric analysis demonstrated that radiation exposure to key organs was well within acceptable safety limits.
The trial also demonstrated a median overall survival of 23 months from initial diagnosis, or 13 months from the initiation
of treatment in the recurring setting. Of the nine patients who received conventional imaging, four (44%) exhibited stable
disease at day 135 and two (22%) at day 180, determined by longitudinal imaging.
The most frequent treatment emergent adverse events ("TEAEs") were decreased lymphocyte count, fatigue, headache
and hiccups, which occurred in three patients (30%), followed by decreased platelet count, diarrhea, cerebral oedema
(swelling), and insomnia, which occurred in two patients (20%). Except for cerebral oedema (swelling), a common
adverse event following radiation to the brain, adverse events were of low grade, did not show any trends or patterns
and were clinically manageable, with a significant proportion deemed unrelated to therapy.
In 2023, we initiated a Phase 1 trial, IPAX-2, to further evaluate the safety of TLX101-Tx in up to 15 patients as a front-line
therapy for the treatment of glioblastoma in combination with EBRT and temozolomide. In IPAX-2, the second patient
cohort has completed and the third cohort is fully enrolled.
TLX101-Tx was investigated in the recurrent setting in the investigator-initiated IPAX-Linz Phase 2 trial, which reported
preliminary results in April 2025. Treatment with TLX101-Tx was well tolerated with no serious adverse events reported.
IPAX-Linz demonstrated encouraging preliminary efficacy data, indicating a median overall survival ("OS") of 12.4 months
from the initiation of treatment with TLX101-Tx, or 32.2 months from initial diagnosis. Five of the eight patients enrolled in
106
IPAX-Linz had unmethylated MGMT promoter status, which is typically associated with poorer outcomes. These results
are consistent with the positive efficacy outcomes generated in the IPAX-1 study. In comparison, recurrent glioblastoma
patients treated with EBRT alone have a reported median survival of 9.9 months from treatment.
Therapy – TLX102-Tx
In our TLX102-Tx (211At astato-L-phenylalanine) program, we are also exploring how phenylalanine, the same LAT1
targeting small molecule substrate used in TLX101-Tx, radiolabeled with an alpha-emitting isotope might deliver a
different therapeutic profile. Astatine-211 is an alpha-emitting radioisotope with comparable halogen chemistry to
Iodine-131 that can cross the blood-brain barrier. TLX102-Tx has demonstrated pre-clinical proof-of-concept and we
believe that TLX102-Tx has the potential to have a favorable efficacy and safety profile in future human clinical trials in
patients with glioblastoma, leptomeningeal disease, and multiple myeloma. Astatine chemistry has been demonstrated,
scaled up and automated, ready for clinical production. Due to comparable target binding and molecular structure, we
expect that data from our existing LAT1 theranostic programs TLX101-Px and TLX101-Tx will complement and inform the
clinical and regulatory development strategy for TLX102-Tx.
In August 2020, TLX102-Tx was granted orphan drug designation from the FDA in the U.S. for the treatment of multiple
myeloma, and in November 2025, TLX102-Tx was granted orphan drug designation from the FDA for the treatment of
malignant gliomas. Orphan drug designation may not lead to a faster development or regulatory review or approval
process in multiple myeloma or glioblastoma and does not increase the likelihood that TLX102-Tx will receive marketing
approval in either of these disease areas.
In December 2025, the FDA provided positive written feedback on our proposed two-part FIH study design in patients
with leptomeningeal disease ("LMD"), and acknowledged that LMD from solid tumors represents a significant unmet
medical need. The FDA has also provided guidance on a combined protocol for imaging and therapy to enable a true
theranostic evaluation, including potential future label expansion for TLX101-Px.
Imaging – TLX101-Px
TLX101-Px (18F-FET) is a radiolabeled amino acid PET agent for imaging of gliomas that is used in clinical research
settings, including in our IPAX series of trials of TLX101-Tx, as a complementary diagnostic agent. Clinical data suggest
that TLX101-Px can facilitate the identification of recurrence of brain metastases. 18F-FET is widely used in many
jurisdictions and is recommended by the joint guidelines from the European Association of Nuclear Medicine, European
Association of Neuro-Oncology, Society of Nuclear Medicine and Molecular Imaging, The European Society for Pediatric
Oncology and The Response Assessment in Pediatric Neuro-Oncology for the characterization of recurrence in glioma
patients. 18F-FET is also included in latest NCCN Guidelines® for Central Nervous System Cancers (V1.2025) for PET
imaging of gliomas.
In October 2020, TLX101-Px was granted orphan drug designation from the FDA in the U.S. for the imaging of glioma.
Orphan drug designation may not lead to a faster development or regulatory review or approval process and does not
increase the likelihood that TLX101-Px will receive marketing approval.
We used TLX101-Px to select patients and track disease response in our IPAX-1 Phase 1/2 clinical trial and the recently
completed IPAX-Linz investigator-initiated trial. We are using TLX101-Px in the Phase 1 IPAX-2 trial, which is actively
dosing patients, and for patient selection in our pivotal IPAX BrIGHT trial, as well as assessing metabolic tumor response
according to PET-based response assessment criteria for diffuse gliomas ("PET RANO 1.0").
In August 2024, we submitted an NDA to the FDA for TLX101-Px for the characterization of progressive or recurrent
glioma in both adult and pediatric patients from treatment related changes through the 505(b)(2) NDA regulatory
pathway. In October 2024, the FDA accepted the NDA, granted priority review and assigned a PDUFA goal date of April
26, 2025. In April 2025, the FDA issued a CRL, stating that additional confirmatory clinical evidence was required to
progress the application. Following engagement with the FDA, including a successful Type A meeting, in September 2025
we reached agreement regarding the required resubmission package, which includes a retrospective analysis to
supplement clinical data. We are currently finalizing our resubmission package and will provide an update upon filing.
There can be no assurance that the FDA will accept our resubmitted NDA for review, or approve TLX101-Px. TLX101-Px
was granted fast track designation by the FDA for this indication in April 2024. Fast track designation may not lead to a
faster development or regulatory review or approval process, and does not increase the likelihood that TLX101-Px will
receive marketing approval.
In February 2026 we submitted a MAA for TLX101-Px in a selection of European countries, with France acting as RMS.
The French National Agency for Medicines and Health Products Safety, ANSM, in its capacity as RMS is responsible for
coordinating and leading the scientific evaluation of the dossier, in collaboration with the Concerned Member States.
There can be no assurance that the MAA will be validated, or that marketing authorizations for TLX101‑Px will ultimately
be granted.
We also intend to conduct a label-expanding Phase 3 trial of TLX101-Px for the imaging of patients with brain metastases
from non-brain cancers, including lung and breast cancer.
107
The key attributes supporting development of TLX101-Px include:
•Potential tool for management of progression and treatment monitoring;
•Orphan drug designation, potential to meet major unmet need; and
•Widely used in Europe and recommended in the joint guidelines for imaging of gliomas.
Clinical Data
The ability of 18F-FET PET to characterize gliomas across tumor grades and clinical settings has been evaluated in
multiple prospective and retrospective clinical studies (Dunet et al. J Nucl Med. 2012). Various imaging techniques were
used to compare 18F-FET PET with magnetic resonance imaging ("MRI"), 18F-fluoro-2-deoxy-D-glucose ("FDG-PET"), 18F-
fluorothymidine, and perfusion-weighted MRI (Juhász et al. Mol Imaging. 2014). The findings from these trials indicated
that 18F-FET PET generally demonstrated higher sensitivity and specificity (Galldiks et al. EJNMMI. 2015; Galldiks et al.
Neuro Oncol. 2015; Albert et al. EJNMMI. 2016). 18F-FET was the subject of a systematic review and meta-analysis
(Dunet et al. J Nucl Med. 2012) that included multiple studies and patient lesions, concluding that 18F-FET PET may serve
as a complementary modality to standard of care MRI in managing brain malignancies.
As part of our commitment to provide access to medicine, we are running an EAP in the U.S. to allow access to TLX101-
Px outside of a clinical trial to patients for whom there are no comparable or satisfactory alternative options.
We are also exploring applications of TLX101-Px imaging in radiation treatment planning through the 18F-FET in
glioblastoma ("FIG") investigator-initiated trial. This aim is to show that TLX101-Px can help improve radiation treatment
planning in a prospective, multi-center PET/CT trial.
Our Other Solid Tumors and Hematologic Oncology Programs
Soft Tissue Sarcoma and PDGFRα
Soft tissue sarcoma ("STS") is a rare, complex disease that encompasses a diverse group of relatively rare cancers, with
more than 50 histological subtypes. According to the National Cancer Institute, there were an estimated 13,400 new
cases and 5,140 deaths were caused by STS in 2023 in the U.S. Standard treatment for STS includes surgery, radiation
therapy and/or chemotherapy. For patients with advanced, unresectable, or metastatic disease, treatment typically
involves chemotherapy with single agents (e.g., doxorubicin) or anthracycline-based combination regimens. However,
the prognosis for these patients remains poor, with treated patients with metastatic disease having a median overall
survival of around 12–18 months.
STS is usually diagnosed using imaging tests (CT, MRI and/or FDG-PET) and/or biopsy, depending on the tumor location.
Conventional imaging and biopsy are also used for staging.
There are several programs in clinical development for the treatment of STS, none of which is a targeted systemic
radiotherapy. We consider our most direct competitors to be companies developing systemic radiotherapies in the soft-
tissue sarcoma space, including OncoTherapy Science, RadioPharm Theranostics and Cellectar Biosciences, Inc. Our
competitors will also include companies developing other modalities to treat STS.
Therapy – TLX300-Tx
In April 2022, we entered into a licensing agreement with Lilly that granted us exclusive worldwide rights to develop and
commercialize radiolabeled forms of olaratumab as a targeting agent for radiopharmaceutical imaging and therapy of
cancer. Lilly originally developed olaratumab a non-radiolabeled monoclonal antibody targeting PDGFRα, a protein
expressed in multiple tumor types that is involved in fibrogenesis. Olaratumab has a well-established clinical and
toxicology profile as a non-radiolabeled agent.
Olaratumab was granted accelerated approval in the U.S. and conditional approval in the European Union based on
Phase 2 trial data. Lilly began marketing olaratumab as Lartruvo in 2016.
Sales of Lartruvo peaked at US$304.7 million in 2018. Olaratumab was voluntarily withdrawn from the market by Lilly
following the failure of the Phase 3 ANNOUNCE clinical trial, in which olaratumab in combination with standard
chemotherapy did not improve survival for patients compared to chemotherapy alone. We believe that the therapeutic
limitations of Lartruvo can be overcome through the re-purposing of olaratumab as a radiopharmaceutical.
Our initial development focus for radiolabeled olaratumab is on advanced or metastatic STS and other PDGFRα positive
tumors. Our therapeutic product candidate, TLX300-Tx, employs antibody-directed targeted radiation against PDGFRα.
We believe that the ability of olaratumab to target PDGFRα makes it a promising candidate for use as a targeted
radionuclide therapy.
We completed pre-clinical validation of TLX300-Tx in 2023 and initiated a FIH Phase 1 proof-of-concept targeting and
biodistribution trial, ZOLAR, in April 2025. The aim of the trial is to evaluate the safety, pharmacokinetics, biodistribution
and dosimetry, and establish the optimal dose of TLX300-Px using PET imaging, prior to therapeutic studies, based on a
108
theranostic approach. Patient dosimetry and target expression characteristics from ZOLAR will inform selection of the
therapeutic radionuclide for TLX300-Tx, in conjunction with currently active non-clinical radiation biology studies.
The key attributes supporting development of TLX300-Tx include:
•Well-established clinical and toxicology profile of olaratumab as a non-radiolabeled agent;
•First-in-human, Phase 1 proof-of-concept and biodistribution trial dosing patients in Australia; and
•Potential application in STS and a range of other cancers (e.g., bone, brain, breast, lung, ovarian and prostate
cancers).
In a preclinical study of a dose of 10 MBq of TLX300-Tx in mice, we observed a significant increase in survival to tumor
endpoint (P=0.0004, Log-Rank test).
Imaging – TLX300-Px
TLX300-Px (89Zr-DFOsq-olaratumab) is an investigational imaging agent that we are developing for use with TLX300-Tx
as a theranostic pair. DFO-squaramide ("DFOsq") is our proprietary chelator technology, designed to optimize the
bioconjugate manufacture, conjugate stability and serum stability for use with this isotope. If approved, TLX300-Px
would be the first imaging agent to specifically detect the presence of PDGFRα in patients with STS or other PDGFRα
positive tumors.
Following the completion of pre-clinical studies, we are conducting a first-in-human Phase 1 proof-of-concept targeting
and biodistribution trial in Australia using TLX300-Px. This dose-finding study is assessing safety, tolerability, dosimetry,
pharmacokinetics and imaging properties of TLX300-Px in participants with STS and other PDGFRα-positive tumors,
prior to therapeutic studies. We have not yet determined the specific isotope(s) that we will use in therapeutic trials.
The pre-clinical studies of radiolabeled-olaratumab have demonstrated that olaratumab can be bioconjugated with
chelators and radiolabeled with imaging and therapeutic radionuclides. In a biodistribution study of TLX300-Px in mice
tumor targeting reached 55% of ID/g at 120 hours post-injection, accompanied by accumulation in main clearance organs
as predicted based on radiolabeled antibody clearance. We believe results of these pre-clinical studies demonstrate the
viability of radiolabeling olaratumab, high uptake of the imaging agent in tumors and subsequent clearance and
demonstrated anti-tumor activity with the therapy agent. Animal or pre-clinical results should be interpreted with caution
as they may not correlate to results in human clinical trials.
Hematologic Oncology and CD66
Hematopoietic stem cell transplantation ("HSCT") is an important lifesaving treatment opportunity for various
hematological malignancies and a variety of non-malignant conditions such as severe aplastic anemia, inherited bone
marrow failure syndromes, sickle cell disease, transfusion-dependent thalassemia, inherited immune deficiency
syndromes, and certain metabolic disorders. Experimentally, HSCT has been used in severe refractory autoimmune
diseases.
Conditions such as acute myeloid leukemia, multiple myeloma and systemic amyloid light chain amyloidosis may also
benefit from more tolerable bone marrow conditioning regimens. The utilization of novel cell and gene therapies may
increase by replacing toxic chemotherapy conditioning approaches with bone marrow conditioning.
Our hematologic oncology program targets distinct members of CD66, a family of receptors expressed on specific types
of immune or blood cells that serve as attractive biomarkers for novel experimental conditioning radiopharmaceuticals.
Market and Opportunity for Bone Marrow Conditioning Treatment
According to the World Wide Network of Bone and Marrow Transplantation, there were approximately 90,000 first HSCT
performed in 2019, of which 47% were allogeneic. According to the U.S. Health Resources and Services Administration,
there were approximately 22,000 HSCT performed in the U.S. in 2020, 41% of which were allogeneic.
Prior to undergoing HSCT for the treatment of hematologic malignancies patients undergo a bone marrow conditioning
treatment. Current standard of care typically requires bone marrow conditioning with multi-drug chemotherapy regimens.
However, these regimens are highly toxic, and patients may not tolerate treatment. This creates an important unmet
medical need for more tolerable bone marrow conditioning regimens.
There are several systemic radiotherapies being evaluated in clinical trials as conditioning agents for HSCT. We consider
our most direct competitors to be companies developing systemic radiotherapies in the hematology space, including
Actinium Pharmaceuticals, Inc., Acrotech Biopharma, Inc., Cellectar Biosciences, Inc. and Jasper Therapeutics, Inc.
109
Market and Opportunity for Imaging of Bone Marrow Infection (Osteomyelitis)
The incidence of osteomyelitis is estimated to be as high as 21.8 cases per 100,000 persons per year. The diagnosis of
osteomyelitis is a challenge for diagnostic imaging and timely identification/localization of pathology can be of critical
importance for appropriate management of patients.
Imaging modalities used to diagnose osteomyelitis can include X-ray, bone scintigraphy, CT, and MRI. These are typically
combined with imaging of white blood cells to distinguish infection, sterile inflammation, and other disorders. White blood
cell imaging is typically performed using in vitro separation and labeling of white blood cells, which requires preparation
time and carries the inherent risk of contamination.
Therapy – TLX66-Tx
TLX66-Tx (90Y-DTPA-besilesomab), is a product candidate for bone marrow conditioning for HSCT conditioning, a broad
clinical indication.
Our HSCT conditioning agent, TLX66-Tx, has been evaluated in acute myeloid leukemia, multiple myeloma and systemic
amyloid light chain amyloidosis through investigator-initiated trials. Clinical data suggest TLX66-Tx could be a well-
tolerated (and therefore highly versatile) bone marrow conditioning agent which could be utilized as a single agent or in
combination with either reduced or high intensity conditioning agents preceding both autologous or allogeneic HSCT.
Orchard et al. (Bone Marrow Transplantation. 2024) reported that “All patients engrafted, treatment-related mortality 1-
year post-transplant was zero. Toxicities were no greater than those anticipated for similar conditioning regimens
without targeted radiation. The ability to substantially intensify conditioning prior to haematopoietic stem cell
transplantation without increasing toxicity warrants further testing to determine efficacy.”
TLX66-Tx is currently being studied in a Phase 2 IIT in pediatric high-risk leukemia and we plan to evaluate TLX66-Tx in
a Phase 2 clinical trial as a BMC agent in patients with acute myeloid leukemia who are not suitable for conventional BMC
regimes.
TLX66-Tx was granted orphan drug designation status in the U.S. and Europe. Orphan drug designation may not lead to
a faster development or regulatory review or approval process and does not increase the likelihood that TLX66-Tx will
receive marketing approval.
The key attributes supporting the development of TLX66-Tx include:
•Minimal uptake in non-hematopoietic organs such as liver, kidneys and gut;
•Approximately 80 patients treated in several Phase 1 and 2 investigator-initiated trials of TLX66-Tx in different
hematological diseases requiring autologous or allogeneic stem cell transplantation; and
•Orphan drug designation granted in the U.S. and Europe for TLX66-Tx for bone marrow conditioning.
Manufacturing TLX66-Tx and TLX66-Px utilizes a small amount of Triton X-100, which is a non-ionic surfactant, in the
antibody manufacturing process. Triton X-100 is subject to a regulation in the European Union known as Registration,
Evaluation, Authorization and Restriction of Chemicals ("REACH"). We are permitted to manufacture TLX66-Tx for
research and clinical development in the European Union pursuant to a self-certified exemption applicable to research
and development activity. We would need to obtain authorization under REACH in order to use Triton X-100 for the future
commercial manufacturing of TLX66-Tx or re-design the commercial manufacturing process for TLX66-Tx such that
Triton X-100 is not used. We are currently planning to re-design the commercial manufacturing process for TLX66-Tx
and potentially for TLX66-Px. We believe that any improvements to the manufacturing process we may make could also
result in an increase in productivity and a potential reduction in manufacturing costs. If we re-design the manufacturing
process for TLX66-Tx, we may be required to conduct additional clinical trials of TLX66-Tx or meet alternative regulatory
standards.
Clinical Data
TLX66-Tx has been evaluated in over 80 patients in several IITs as a conditioning agent preceding HSCT in patients with
a range of hematological malignancies, including a Phase 1 dose-escalation trial in 55 patients with hematological
malignancies, a Phase 1 trial in nine patients with pediatric relapsed/refractory leukemia, a Phase 1/2a trial in nine
patients with AL-amyloidosis and a Phase 2 randomized controlled trial in 25 patients with multiple myeloma. In these
trials, there have not been significant toxicities and there have not been detectable non-hematological toxicities such as
mucositis/colitis. In the pediatric population, TLX66-Tx has been well tolerated with no serious toxicities.
In a Phase 2 trial using TLX66-Tx and HD-melphalan in 24 patients as a conditioning agent for multiple myeloma
autologous HSCT, the complete response rate in the combination cohort (12 patients) was 50%, compared to 25% in the
HD-melphalan control group (12 patients).
In reported data from 30 patients out of 55 patients treated in a Phase 1 trial of TLX66-Tx, patients were given increasing
doses of TLX66-Tx followed by reduced intensity conditioning and HSCT. The overall survival rate was 73% ten years
after the HSCT procedure with low toxicity for TLX66-Tx. There were no severe non-hematological adverse events
110
detected and efficient myeloablation, both in bone marrow and peripheral blood (the anticipated therapeutic effect and
prerequisite for both successful autologous and allogeneic HSCT), was observed.
The Phase 1/2a trial evaluating TLX66-Tx in nine patients with AL amyloidosis evaluated the safety and toxicity of
TLX66-Tx as a bone marrow conditioning agent prior to HSCT. All nine patients were successfully engrafted following
bone marrow conditioning with TLX66-Tx and autologous HSCT without any chemotherapy. TLX66-Tx was well tolerated
by all patients and had a very low toxicity profile when compared to chemotherapy-based conditioning regimes. There
were no serious adverse events or transplant-related deaths.
TLX66-Tx is currently being evaluated in a Phase 2 IIT in high-risk pediatric leukemia and we also plan to develop TLX66-
Tx as a bone marrow conditioning agent in high-risk acute myeloid leukemia patients in complete remission with minimal
residual disease in combination with reduced intensity conditioning preceding allogenic HSCT.
Imaging – TLX66-Px
TLX66-Px (99mTc-besilesomab) is our commercial imaging agent for osteomyelitis, approved in 32 European countries
and Mexico (marketed as Scintimun). Scintimun was previously manufactured and distributed by Curium Pharma through
an out-license from Telix via the acquisition of TheraPharm in 2020. Curium Pharma received marketing authorization for
Scintimun in the European Union in 2010 for scintigraphic imaging, in conjunction with other modalities, for determining
the location of inflammation/infection in peripheral bone in adults with suspected osteomyelitis. Following a strategic
review of the asset, we elected to bring sales and marketing in-house in 2025. TLX66-Px has not received marketing
approval in the U.S. We are evaluating the feasibility of filing for a marketing authorization application in the U.S. where
we retain the rights.
The key attributes supporting the use of TLX66-Px include:
•EMA approval for imaging of peripheral osteomyelitis in 2010; and
•Phase 3 trial showed that Scintimun imaging is accurate and well-tolerated in diagnosing infection of the peripheral
skeleton and provides comparable information (Richter et al. EJNMMI. 2011).
Clinical Data
The approval of Scintimun was based on the results of a multicenter study performed in 22 European centers. This
multinational, Phase 3 clinical study was undertaken to compare anti-granulocyte imaging using Scintimun with 99mTc-
labelled white blood cells in patients with peripheral osteomyelitis. The results of this Phase 3 trial showed that Scintimun
imaging is accurate and well-tolerated in diagnosing infection of the peripheral skeleton and provides comparable
information to 99mTc-labelled white blood cells in patients with chronic osteomyelitis.
MedTech
Our MedTech business is advancing surgical solutions and digital products that power our precision medicines and
therapeutics across the entire patient journey from diagnosis to surgical intervention and therapy. We anticipate applying
this first in urology, for prostate and kidney cancer, and then across the breadth of indications we are pursuing.
Radio-Guided Surgery ("RGS")
Bringing molecular imaging into the operating theater is a key part of our portfolio strategy for urologic oncology.
In November 2023 we acquired the SENSEI radio-guided surgery business from Lightpoint Medical Ltd ("Lightpoint").
SENSEI is a miniature gamma probe device used to detect radiation in patients and guide surgery. The probe is inserted
into a surgical port and can then be controlled by the clinician during the procedure. When used with targeted imaging
agents, SENSEI may enable the intraoperative detection of cancer in real time, supporting greater precision in the
removal of tumors.
The utility of SENSEI has been demonstrated in several studies. These include a prospective multicenter trial assessing
the safety and performance of the SENSEI probe for prostate cancer sentinel lymph node biopsy. The primary objective
was the sentinel lymph node dissection rate with a 100% detection rate achieved by the drop-in probe and no adverse
events linked to the probe. The study concluded that the SENSEI probe meets performance and safety requirements for
sentinel lymph node biopsy in prostate cancer, offering improved maneuverability and sentinel lymph node detection
compared to the conventional rigid laparoscopic gamma probe. Another study covering ten patients concluded that using
the probe is also safe and feasible for sentinel lymph node detection in early-stage cervical cancer. Ongoing studies in
the U.S. (Weiner et al. European Urology Open Science. 2024) and Germany (Falkenbach et al. Current Opinion in
Urology. 2025; Harke et al. Clin Nucl Med. 2024; Bravi et al. Eur Urol Focus. 2025) are using a PSMA-targeted
radiotracer in conjunction with the SENSEI drop-in probe to detect and remove metastatic lymph nodes. One study
reported successful removal in 88% of patients, with 71% achieving complete biochemical response (PSA < 0.2 ng/mL). A
multicenter analysis across 11 sites (259 men) found that PSMA-radio-guided surgery led to lower postoperative PSA
levels, longer time to follow-up (7 vs 24 months), and higher two-year cancer control rates (18% vs 30% BCR-free
survival, 51% vs 73% CR-free survival) compared to standard surgery. These findings suggest PSMA-radio-guided
surgery offers more precise treatment and better short-term outcomes for prostate cancer patients with nodal
recurrence, with longer term oncological benefit still under investigation. SENSEI has a Conformité Européenne ("CE")
111
mark for sentinel lymph node biopsy applications in prostate, endometrial and cervical cancers, and is registered with the
FDA as a class I, 510(k) exempt medical device.
In September 2025, we announced a collaboration with IMRA Surgical, an Australian-based company at the forefront of
disruptive technology for surgical training solutions, to accelerate the development of advanced training models and
enhance the integration of cutting-edge radio-guided surgical technologies. IMRA Surgical has established itself as a
leader in the development of synthetic surgical phantoms, medical models made from hydrogel that accurately simulate
the response of actual human tissue. These models are transforming surgical training by enabling trainees to practice
complex procedures in nearly lifelike conditions, without relying on cadavers or animal tissue. The company’s innovative
models are designed to reduce the learning curve for surgeons, ensuring that they gain hands-on experience with
greater precision and efficiency, ultimately leading to fewer mistakes and better patient outcomes. As part of the
collaboration, IMRA Surgical will develop a groundbreaking new hydrogel model designed specifically to support
education and training in the use of Telix’s SENSEI drop-in technology. This hydrogel model will be the first of its kind,
providing practitioners with a realistic, hands-on training experience and facilitating a faster learning curve when
adopting advanced radio-guided technology.
Through the IRiS (Imaging and Robotics in Surgery) Alliance with Mauna Kea Technologies ("Mauna Kea"), a leading
medical device company pioneering the development of real-time intraoperative visualization of cancer tissue during
surgery, we are exploring new hybrid pharmaceutical-device products through the combination of our cancer-targeting
agents with Cellvizio, Mauna Kea’s confocal surgical laser endomicroscopy in vivo cellular imaging platform.
Dosimetry
In April 2024, we entered into an agreement to commercially partner the QDOSE dosimetry software platform with ABX-
CRO and its development partner, Quantinm AB. QDOSE is a 510(k) cleared, CE-marked and Korean-FDA approved
software platform designed to enable reliable estimation of patient-specific dosimetry for both therapeutic and
diagnostic radiopharmaceuticals, supporting the safe and effective integration into clinical practice. We believe that
personalized targeted radionuclide therapy administration based on individual patient dosimetry has the potential to
improve clinical outcomes by optimizing treatment response while reducing effects on normal healthy organs and
optimizing the use of isotope supply chains.
Telix holds the intellectual property rights and leads development of QDOSE. Together with our distributors that serve as
manufacturer and customer support providers, we aim to advance dosimetry and make it more accessible, reliable, and
impactful for healthcare providers.
Artificial Intelligence (AI)
Imaging using targeted radiation relies heavily on digital data processing and input from highly skilled and trained
technicians and radiologists to correctly interpret the data. We believe that AI technology can recognize complex
patterns in large datasets and conduct predictive analysis, with potential to transform imaging analysis and improve the
accuracy of decision making for clinicians.
In 2023, we acquired Dedicaid GmbH and its AI platform capable of rapidly generating indication specific clinical decision
support software ("CDSS") applications from available datasets, for use with PSMA-PET amongst other imaging
modalities. Each CDSS application will be trained to predict outcomes such as the severity of disease, risk to the patient
and/or inform treatment decisions. The platform also houses an automated machine learning engine ("AutoML"). We
believe that this platform is differentiated from commercially-available AI solutions currently used in PSMA-PET imaging,
which are limited to supporting clinicians in the interpretation and reading of images – without a prediction capability. The
platform is designed to reduce the time, cost and level of expertise required to build, test and validate new CDSS
applications, facilitating a streamlined development and regulatory pathway for each new application. We are conducting
final validation of the Dedicaid platform.
The Dedicaid acquisition also included a lead medical device tool that is designed to interpret the risk of prostate cancer
advancement from a PSMA-PET scan image by correlating it to a well-known histopathology indicator (the Gleason
Grade). A second AI asset supporting Illuccix is designed to automate the identification and classification of prostate
cancer lesions from PSMA-PET scans to support greater efficiency and standardization in the imaging workflow.
Our focus for the AI platform is to develop AI-powered solutions that support our product candidates and to enable these
solutions for use by the nuclear medicine community as approved medical devices. We aim to use AI across our
development pipeline by utilizing clinical imaging and outcome data as they become available and to develop and
validate medical device applications supporting approved products.
Research and Development
Telix continues to diversify its drug pipeline through the advancement of a new generation of proprietary targeted
biologics and with the expansion of its protein engineering and discovery research facility in Los Angeles, CA. Efforts
underway are synergistically adding several early‑stage drug candidates against high‑value targets, including DLL3 and
integrin αvβ6, enabling expansion into therapy areas with unmet clinical need. This platform of biologics is based on
small, engineered antibody formats that enable highly specific cancer targeting, combined with rapid tumor uptake and
blood clearance. These features allow for more directed payload delivery, helping to minimize off‑target therapeutic side
112
effects. This technology has the potential to be highly effective for imaging and treating tumors with a broad range of
radioisotopes, with diagnostic and therapeutic alpha‑ and beta‑emitters of particular interest.
In addition to developing targeting agents, Telix is committed to utilizing isotopes with the most appropriate physical
characteristics to help deliver the best possible treatments and, ultimately, improved patient outcomes. To support this,
the company is continuously exploring new ways to produce and source these critical components of our drugs. One
emerging alpha‑emitter with significant promise is lead‑212 (212Pb), an isotope to which Telix has steadily increased
access through the design and scale‑up of a thorium‑228 (228Th)–based generator. Telix is preparing to introduce a fully
automated, high‑output generator with a small, single hot‑cell footprint, sufficient 212Pb production capacity for multiple
clinical doses per elution, and the potential to scale based on demand. This production footprint is designed for
deployment throughout Telix’s manufacturing and distribution networks, including the recently acquired RLS
Radiopharmacies network. This development opens new opportunities for the creation of alpha‑emitting
radiopharmaceuticals with a simplified production infrastructure compared with other alpha‑emitting radioisotopes. The
alpha‑emitting profile of 212Pb also offers practical synergies with the engineered antibodies described above.
Telix Manufacturing Solutions (TMS)
We are focused on enhancing our existing global manufacturing and supply chain with a balance of external and in-house
capabilities, securing a robust and innovative manufacturing infrastructure and supply chain to serve our patients.
Manufacturing and supply chain supporting our portfolio broadly cover the following areas: radioisotopes,
radiochemistry, biologics, small molecules, fill/finish, packaging and labeling, and storage and distribution.
Since 2022, we have made significant progress with the buildout of our radioisotope manufacturing facility in Brussels
South. We have been granted an updated radiation license by the Belgian Federal Agency for Nuclear Control, enabling
site activation subject to the regulatory inspections and approvals.
Our approximately 30,000 square foot radioisotope manufacturing facility is one of Europe’s largest radiopharmaceutical
production facilities. The site will enable improved access to radiopharmaceuticals for patients across the EMEA region
and the world as a primary GMP-capable manufacturing site for our clinical and Commercial Products. The site also has
extensive R&D capabilities, with a focus on alpha-emitting isotopes. We believe the proximity of an alpha
radiopharmaceutical laboratory to a production GMP environment is a differentiated capability to our competition. We
expect the site to evolve and develop as a hub for strategic collaborations via R&D facilities and manufacturing line
designated for university and SME partners.
We aim to have a degree of vertical integration in our three operating regions. In line with this goal, in 2022 we acquired
Optimal Tracers, a California-based company that provides radiochemistry process development services and research
tracers for use in clinical trials. The acquisition of Optimal Tracers expanded our translational radiochemistry capability
and establishes a U.S.-based laboratory and production footprint for clinical trial doses, now known as TMS Sacramento.
In April 2024, we acquired IsoTherapeutics Group, LLC ("IsoTherapeutics"), a specialty radiopharmaceutical development
and bioconjugation firm, based in Texas. We expect that the acquisition will further enhance our internal drug
development capabilities.
In April 2024, we also acquired ARTMS Inc. ("ARTMS"), a radioisotope production technology company based in Canada,
and its advanced cyclotron-based isotope production platform, manufacturing plant and stockpile of ultra-pure rare
metals required for consumable target production. We expect that the acquisition will further enhance the vertical
integration of our supply chain and manufacturing by providing a greater level of control and security over each of our
diagnostic isotopes.
In January 2025, we acquired RLS (USA) Inc., America’s only Joint Commission-accredited radiopharmacy network
distributing PET, SPECT and therapeutic radiopharmaceuticals. The RLS acquisition augments our existing distribution
network for last-mile delivery and provides expansionary space to build out a radiometal production network to meet
future demand for radiopharmaceuticals.
In June 2025, we established TMS Yokohama, in Yokohama, Japan. Telix’s first cyclotron facility in the Asia Pacific region
represents a significant milestone in our global manufacturing strategy. TMS Yokohama will serve as a hub for
commercial and clinical supply, and future research and development in the region. It expands our global production
network which includes in-house and partner facilities. Originally opened in 2018, the site comprises a cyclotron and
multiple production hot cells and was designed and built by JFE Engineering ("JFE") as the Contract Manufacturing
Organization ("CMO") for TLX250-Px in Japan and China, including for the ZIRCON-CP study.
Assuming operational management of this facility will provide greater control over existing clinical supply with the
possibility to expand production to other Telix investigational and future commercial products in the region, including
Illuccix and Pixclara for Greater Tokyo, and TLX591-Tx for the Asia Pacific region.
During 2025, we progressed the construction of a new facility in Melbourne, Australia, to be known as TMS North
Melbourne, for early-phase clinical research and radiopharmaceutical production, to support APAC translational research
and clinical trials. In September 2025, both TMS North Melbourne and TMS Yokohama were granted radiation licenses
for a broad range of clinically and commercially important medical isotopes.
113
Our biologics, small molecule, aseptic fill/finish and commercial secondary packaging manufacturing and supply chain are
accomplished through relationships with external contract manufacturing and development organizations ("CDMOs"). We
have agreements with ABX-CRO, Abzena Holdings (U.S.) LLC., Curia Global, Dalton, DiverChim CDMO, Eurofins,
GenScript ProBio, Goodwin Biotechnology Inc., Grand Rapids Aseptic Manufacturing, Patheon Pharma Services, PCI
Pharma Services, Sharp Packaging, Sharp Sterile Manufacturing, and UPS. We are also pursuing the addition of in-house
capabilities (secondary packaging, antibody conjugation, and fill/finish) where appropriate through vertical integration.
With respect to producing radiolabeled drug product, we aim to continue to deepen our relationship with key
manufacturing networks in the U.S.: PharmaLogic for 18F and 89Zr products, and Cardinal Health for 68Ga and 89Zr and
225Ac products. We have agreements with Evergreen Theragnostics, AtomVie Global Radiopharma, Eckert & Ziegler SE,
Seibersdorf Laboratories and South Australian Health and Medical Research Institute for the manufacture of our
therapeutic product candidates across multiple regions, and we are working on establishing additional key manufacturers
in APAC and the European Union. Our current capabilities encompass products radiolabelled with 177Lu, 131I, and 89Zr, and
we aim to build-up our capabilities with respect to producing products radiolabelled with alpha-emitters such as 225Ac
and 212Pb.
We are dedicated to enhancing our global supply chain capabilities, particularly for the clinical and commercial supply of
isotopes used in radiolabeling, as well as for supplying generators. We have established a series of strategic supply
agreements with leading industry partners to supply starting material for internal production as well as the final
radioisotope used in radiolabeling. These partnerships include the Australian Nuclear Science and Technology
Organisation, Cardinal Health, Eckert & Ziegler SE, Isotopia, ITM, PanTera, Radnostix, SHINE Technologies, Thor Medical,
and Van Overeem Nuclear, and are pivotal in ensuring a broad and robust supply network for both therapeutic and
diagnostic isotopes. By diversifying our supply chain through these contracts, we aim to create a resilient system that
eliminates dependencies on a single supply chain. This approach is intended to ensure uninterrupted supply and to
enhance our capability to meet growing demand.
Through these strategic agreements, we aim to maximize the available production process methods and diversify the
reactors used for irradiation and production. This not only ensures a steady and diverse supply but also allows us to
adapt quickly to changing market demands and regulatory environments.
We aim to actively pursue the development and supply of future isotopes. Understanding the critical role these materials
play in advancing medical and scientific endeavors, we are dedicated to ensuring a robust and resilient supply chain that
can adapt to the evolving needs of the industry.
Our approach is multi-faceted, focusing on strategic partnerships, technological innovation, and sustainable practices.
We continuously seek to expand our network of suppliers and collaborators, forming alliances with leading entities in the
field. This not only diversifies our supply sources but also fosters innovation through shared expertise and resources.
Moreover, we are investing in cutting-edge technologies and processes that enhance our production capabilities,
ensuring efficiency and reliability. Our commitment to sustainability, particularly in the recycling of materials, further
strengthens our supply chain, reducing environmental impact while maximizing resource utilization.
We recognize that the future of isotope supply lies in our ability to anticipate and respond to market changes and
scientific advancements. Therefore, we are dedicated to ongoing research and development, ensuring that we remain at
the forefront of isotope supply. Our goal is not just to meet current demands but to be a driving force in the development
of new isotopes, paving the way for groundbreaking applications that can transform industries and improve lives.
Our commitment to a robust and resilient supply chain for future isotopes is unwavering. We understand the significance
of our role in this dynamic field and are dedicated to maintaining the highest standards of quality, reliability, and
innovation in all our endeavors.
Through these comprehensive efforts, we are seeking to position ourselves as a leader in the supply of isotopes for
radiolabeling, backed by a supply chain that is as diverse as it is robust, ensuring the highest standards of quality and
reliability for our clients.
Sales and Marketing Operations
Our commercial operations span the Americas, EMEA, and APAC Regions. Illuccix is approved in the U.S., Australia, Brazil,
Canada, the United Kingdom, 19 European Economic Area member states, and permitted to be sold in New Zealand, and
we are commercializing this product in these countries through local sales forces, which currently include over 40
associates, and together with distributor partners. Gozellix is approved in the U.S. We have secured a number of
commercial partnerships covering certain geographies to enable distribution and/or commercialization of our products.
In the U.S., we have established a commercial radiopharmacy network of over 225 commercial radiopharmacies to
distribute Illuccix, including partnerships with Cardinal Health, Inc., PharmaLogic Holdings, Corp., and Jubilant Pharma
Ltd. We also have a distribution agreement with Isologic Innovative Radiopharmaceuticals Ltd for the Canadian market.
In Asia Pacific, we have secured a strategic collaboration with Grand Pharmaceutical Group Limited ("Grand Pharma") in
the Greater China area including Mainland China, Taiwan, Hong Kong and Macau. Grand Pharma has been appointed as
our partner for this territory with exclusive development and commercialization rights to our portfolio.
114
In Europe, we have exclusive distribution agreements for the upcoming launch of Illuccix in a number of geographies,
including with Eckert & Ziegler SE in Germany, Xiel Ltd in the United Kingdom, IRE Elit S.A. in France, Radius S.r.l. in Italy,
Biokosmos S.A. in Greece and Cyprus, Sociedade Avanço, Unipessoal, Lda in Portugal, THP Medical Products Vertriebs
GmbH in Austria, Czech Republic and Slovak Republic and WIIK Pharma ApS in Denmark, Finland, Norway and Sweden.
Competition
Our potential competitors include all entities developing and commercializing diagnostics and therapies in the field of
oncology, through nuclear medicine and other modalities. This includes companies, academic institutions, government
agencies, hospitals, other organizations involved in research, manufacturing, and commercialization of diagnostics and
therapies. In addition to the current standard of care for patients, commercial and academic clinical trials are being
pursued by a number of parties in the field of radiopharmaceuticals. Early results from these trials have fueled continued
interest in radiopharmaceuticals, which is being pursued by several biotechnology companies, as well as by large
pharmaceutical companies.
There are several companies with approved beta-based radiopharmaceuticals, including Novartis AG, Bayer AG, Sirtex
Medical, Inc., Boston Scientific Corporation, Acrotech Biopharma LLC and Q BioMed Inc. and other companies developing
beta-based radiopharmaceuticals, including Lantheus Holdings, Inc., Eli Lilly & Company Ltd., ITM Isotope Technologies
Munich SE, Curium Holding France S.A.S, Clarity Pharmaceuticals Limited, The Bracco Group (through its Blue Earth
Therapeutics Ltd. Subsidiary), and Y-mAbs Therapeutics, Inc. The beta emitting isotopes used by these companies
include iodine-131, lutetium-177, strontium-89, copper-67, and yttrium-90.
There are several companies developing targeted alpha-based radiopharmaceuticals for the treatment of cancer,
including Bayer AG, Novartis AG, AstraZeneca PLC/Fusion Pharmaceuticals Inc., Alpha Tau Medical, ARTBIO, Inc., Orano
Med SAS, Johnson & Johnson, Abdera Therapeutics, Inc., Actinium Pharmaceuticals, Inc, Aktis Oncology, Inc.,
Convergent Therapeutics, Inc., ITM Isotope Technologies Munich SE, Perspective Therapeutics, Inc., Eli Lilly & Company
Ltd., RadioMedix, Inc., Bristol Myers Squibb Company and Y-mAbs Therapeutics, Inc. The only approved alpha particle-
based therapy is Bayer’s Xofigo (Radium-223) which was approved in 2013 for the treatment of prostate cancer with
symptomatic bone metastases.
We consider our most direct competitors to be companies developing and commercializing diagnostics and therapies in
our core therapy areas, including prostate cancer, kidney cancer, bladder cancer, brain cancer, sarcoma, and
hematology.
In prostate cancer therapy, Pluvicto (lutetium (177Lu) vipivotide tetraxetan), marketed by Novartis AG, was approved by
the FDA for the treatment of patients with PSMA-positive mCRPC who have been treated with androgen receptor
pathway inhibition and taxane-based chemotherapy in March 2022. In March 2025, Pluvicto was approved by the FDA
for earlier use prior to chemotherapy, in PSMA-positive mCRPC. Pluvicto is the only FDA-approved PSMA-targeted
therapy for the treatment of prostate cancer. Several other systemic radiotherapies are being investigated in clinical
trials in the mCRPC setting and across other stages of prostate cancer, and potentially could be commercialized in the
future.
In mCRPC treatment, there are several companies developing PSMA-targeted therapies in the mCRPC space, including
Novartis AG, Convergent Therapeutics, Inc., Eli Lilly & Company Ltd., Lantheus Holdings, Inc, Curium Holding France
S.A.S, ARTBIO, Inc., The Bracco Group (through its Blue Earth Therapeutics Ltd. subsidiary), Clarity Pharmaceuticals Ltd.,
AstraZeneca PLC, Bayer AG, Orano Med SAS, Isotopia Molecular Imaging Ltd, ITM Isotope Technologies Munich SE,
Johnson & Johnson, AdvanCell Isotopes Pty Ltd, Alpha-9 Theranostics, Inc., Cancer Targeted Technology, LLC,
FutureChem Co Ltd., Sinotau Pharmaceutical Group, Norroy Biosciences Co. Ltd., RadioPharm Theranostics Limited,
Precision Molecular, Inc., CellBion Co., Ltd., StarPharma Holding Limited, Amgen Inc., Crescendo Biologics Limited,
Poseida Therapeutics, Inc., Janux Therapeutics, Inc., Vir Therapeutics, Inc., Bivision Pharmaceuticals, Inc., GlyTherix Ltd,
Jiangsu Hengrui Pharmaceuticals Co., Ltd and Full-Life Technologies Limited. Our competitors also include companies
developing other modalities to treat patients with mCRPC.
In prostate cancer imaging, UCLA and UCSF obtained FDA approval for 68Ga-PSMA-11 in 2020, this was the first PSMA-
PET imaging agent to be approved by the FDA. Pylarify (18F-piflufolastat), marketed by Lantheus Holdings, Inc, was
approved by the FDA in 2021. Locametz (68Ga-PSMA-11), marketed by Novartis, received FDA approval in 2022 and
Posluma (18F-flotufolastat), marketed by The Bracco Group (through its Blue Earth Diagnostics Ltd. subsidiary), received
FDA approval in 2023. Several other PSMA-PET product candidates are being evaluated in clinical trials for prostate
cancer imaging and may be commercialized in the future. Companies developing PSMA-PET imaging agents include
Curium Holding France S.A.S., Clarity Pharmaceuticals Limited, ABX advanced biochemical compounds GmbH, Isotopia
Molecular Imaging Ltd, Itel Telecomunicazioni Srl, ITM Isotope Technologies Munich SE, Five Eleven Pharma, Inc.,
RadioMedix, Inc., CellBion Co., Ltd., Norroy Biosciences Co. Ltd., HTA Co. Ltd and Jiangsu Hengrui Pharmaceuticals Co.,
Ltd.
In kidney cancer therapy, there are several companies developing CAIX-targeted systemic radiotherapies, including ITM
Isotope Technologies Munich SE, Precision Molecular, Inc., Norroy Biosciences Co. Ltd., AstraZeneca PLC/Fusion
Pharmaceuticals Inc., PeptiDream Inc., Bayer AG and Bristol Myers Squibb Company. Our competitors also include
companies developing other modalities to treat patients with kidney cancer.
115
In kidney cancer imaging, there are several companies developing ccRCC or CAIX-targeted imaging agents, including
ITM Isotope Technologies Munich SE, Philogen S.p.A., ImaginAb, Inc., Precision Molecular, Astellas Pharma, Inc., Norroy
Biosciences Co. Ltd., PeptiDream Inc., and Five Eleven Pharma, Inc.
In bladder cancer therapy, there are several companies developing systemic radiotherapies, including Aktis Oncology,
Inc., Glytherix Ltd., AstraZeneca PLC and NuView Life Sciences, Inc. Our competitors also include companies developing
other modalities to treat patients with bladder cancer.
In glioblastoma therapy, there are several companies developing systemic radiotherapies for brain tumors, including ITM
Isotope Technologies Munich SE, PeptiDream Inc., Molecular Targeting Technologies, Inc., EvaThera Theranostics,
Novartis AG, RadioPharm Theranostics Limited, Plus Therapeutics, Inc., Ariceum Therapeutics GmbH, Boston Scientific
Corporation, and Cellectar Biosciences, Inc. Our competitors also include companies developing other modalities to treat
patients with glioblastoma.
In brain cancer imaging, there are several companies developing imaging agents for primary brain tumors and brain
metastases, including Novartis AG, The Bracco Group (through its Blue Earth Diagnostics Ltd. subsidiary), RadioPharm
Theranostics Limited, Curasight A/S, Molecular Targeting Technologies, Inc., and BoomRay Pharmaceuticals Co., Ltd.
In sarcoma, there are several companies developing systemic radiotherapies in the soft-tissue sarcoma space, including
OncoTherapy Sciences, Inc., RadioPharm Theranostics Limited, Ratio Therapeutics, Inc., Y-mAbs Therapeutics, Inc., and
Cellectar Biosciences, Inc.
In hematology, there are several companies developing systemic radiotherapies in the hematology space, including
Actinium Pharmaceuticals, Inc., Bayer AG, Sensei Biotherapeutics, Inc., ImaginAb, Inc. Acrotech Biopharma Inc., Nordic
Nanovector ASA, Orano Med SAS, Samus Therapeutics, Inc., Cellectar Biosciences, Inc. and Jasper Therapeutics. Inc.
Many of our current or potential competitors, either alone or with their collaboration partners, have significantly greater
financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical
trials, obtaining regulatory approvals and marketing approved products than we do. Mergers and acquisitions in the
pharmaceutical and biotechnology industries may result in even more resources being concentrated among a smaller
number of our competitors. Smaller or early-stage companies may also prove to be significant competitors, particularly
through collaborative arrangements with large and established companies. These competitors also compete with us in
recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and patient
enrollment in clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs.
We could see a reduction or elimination in our commercial opportunity if our competitors develop and commercialize
drugs that are safer, more effective, have fewer or less severe adverse events, are more convenient to administer, are
less expensive or with a more favorable label than our product candidates. Our competitors also may obtain FDA or other
regulatory approval for their drugs more rapidly than we may obtain approval for ours, which could result in our
competitors establishing a strong market position before we are able to enter the market. The key competitive factors
affecting the success of all of our product candidates, if approved, are likely to be efficacy, safety, convenience, price,
availability of the relevant isotope, the effectiveness of imaging diagnostics, the level of generic competition and the
availability of reimbursement from government and other third-party payors.
Intellectual Property
Overview
Patent Protection
By their very nature, radiopharmaceuticals must be delivered through a complicated supply chain and go-to-market
model requiring specialized physics, chemistry and biological expertise for successful development and
commercialization protected by know-how and trade secrets. This specialization provides a practical barrier to
competitor entry without the same specialist expertise. Additionally, we aim to build, maintain and continuously improve
our exclusivity and patent position to protect our innovation contribution. We aim to integrate regulatory filing strategy
designed to maximize regulatory market or data exclusivity and through targeted patent protection across the spectrum
of compound, dosing, radiolabeling technology, handling, preparation process and manufacturing inventions.
Older radiopharmaceuticals were historically routinely used in the public domain for many years under practice of
pharmacy or individual named patient prescribing regulatory pathways. This has the benefit of established use and real-
life clinical application and experience for such products when made commercially available, but does potentially create
the result that patent protection is not available or has only limited remaining patent term.
Our original third-party licensed products were in-licensed and were accepted on an “as-is” basis. We have limited
opportunity to determine or influence territory and scope of third-party licensor portfolio and the time to make changes
to scope or territory has long since passed under applicable patent laws. However even for these earlier products, we
have expanded our patent portfolio where possible and seek to obtain related new patents for updates in handling,
dosing and manufacturing to maximize patent exclusivity where feasible, in addition to our supply chain know-how and
trade secrets.
116
For our newer programs and next generation radiopharmaceuticals, the patent protection is deeper and wider across the
spectrum of compound, method of treatment, dosing, radiolabeling technology, handling, preparation process and
manufacturing inventions based on our newer proprietary technologies or due to our innovation in the end-to-end
process.
We have in-licensed registered intellectual property associated with our key therapeutic products: TLX591-Tx, TLX250-
Tx, TLX101-Tx, TLX102-Tx, TLX090-Tx, TLX300-Tx, TLX66-Tx, TLX597-Tx, and related imaging product TLX250-Px in
addition to supplementary intellectual property owned by us. Intellectual property for Illuccix, Gozellix, TLX592-Tx,
TLX252-Tx, TLX400-Tx. RHN001-Tx and RHN001-Dx, and TLX101-Px is wholly owned by us. We have also filed our own
applications for registered patents and trademarks in respect of our key products.
Patents are granted by national and regional intellectual property offices in accordance with the corresponding national
laws. Granted patents provide a right to exclude others from making, using, selling, offering to sell, or importing the
invention as set forth in the claims of the patent. Protection is generally limited to actions in or relating to the countries in
which protection is obtained, and enforcement is generally by litigation.
Patents generally have a maximum term of 20 years after its earliest effective filing date, subject to the payment of
renewal fees in all the relevant countries.
In the field of pharmaceuticals, patent term extensions or supplementary protection certificates may extend the term of a
patent beyond 20 years in certain jurisdictions. Examples of important jurisdictions where these regimes are available are
the U.S., Europe, China, Japan and Australia. Many of the patents and patent applications, if granted, that are in-licensed
or owned by us may be able to be extended under the patent term extension or supplementary protection certificate
regimes (in jurisdictions where these regimes are available) once the key products have been the subject of regulatory
approval as the claims are directed to pharmaceutical products and their uses. The extensions in term are typically up to
five years in duration and are often related to the delay between the grant of the patent and regulatory approval of the
pharmaceutical product.
Requirement for Patentability
The requirements for patentability differ in detail from country to country. However, in general terms the main
requirements are that the invention relate to patentable subject matter; that the invention is novel and has an inventive
step; and that the patent contain an adequate disclosure of performing or making the invention. In order to be novel, the
invention must not have been disclosed in writing or otherwise in public, or offered for sale, before the priority date. The
requirement of inventive step is, in general terms, that the invention must go beyond what the skilled worker in the field
would arrive at as a matter of course when attempting to address the same problem as the invention.
Procedure for Obtaining Patent Protection
Patents are granted on a national basis. International patent protection is based upon a system of well-established and
widely adopted international conventions. The first application for a patent for an invention is called the priority
application, and its filing date is known as the priority date. If patent applications having the same specification are filed
within a year from the priority date in other countries, then (in accordance with the Paris Convention, World Trade
Organization ("WTO") Treaty and bilateral agreements) they retain the effective filing date of the priority application for
the purpose of assessing novelty and inventive step.
There are three different types of patent application of relevance. A provisional application acts as a filing to obtain a
priority date. It does not proceed to grant and is not examined by the patent authorities; rather, a later application must
be filed within a year of its priority date to claim the benefit of that filing. A national filing is a regular patent application in
a particular country or region. It will be examined in most cases by the local or regional patent authorities. Applications
can be filed directly in the country or region, or using another convention called the Patent Cooperation Treaty ("PCT").
The PCT allows for a single application to be filed in a single patent office, designating all the member states, obtain a
preliminary search and opinion, and delay filing into the national and regional intellectual property offices for a period of
30 months from the priority date. The PCT currently has 148 members, including all OECD member countries. At the end
of this period, national filings must be made in the countries of interest.
The patent application is examined in each country (or in some cases regional offices) according to its national laws and
procedures.
Potential Limitations of Patent Protection
Certain limitations are inherent in the patent system. In all relevant countries it is possible to challenge the validity of a
patent even after it has been granted by the intellectual property office. This may be possible by administrative
processes at the relevant patent office, court procedures, or both. However, the laws of some jurisdictions do not
protect intellectual property rights in the same manner and to the same extent as laws in the U.S. Consequently, we may
not be able to prevent third parties from practicing our inventions in all jurisdictions outside the U.S. Competitors may
use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and
further, may export otherwise infringing products to territories where we have patent protection, but enforcement of
such patent protection is not as strong as that in the U.S. These products may compete with our products and any then-
117
approved product and our patents or other intellectual property rights may not be effective or sufficient to prevent them
from competing.
A successful challenge to validity will result in the claims of the patent being narrowed in scope, or the patent being
completely revoked. The grant of a patent by a patent office does not mean it is valid. Because of the limited scope of
material searchable by the patent office, compared to the potential to use documents or act before the priority date that
may not have been identified by the patent office to attack validity, there is a risk that presently unknown disclosures or
actions relevant to patentability will be discovered at a later time, with consequent risks to validity. The scope of a
granted patent may be significantly different to a pending application, and so it is not possible to advise with certainty in
relation to infringement of a pending application.
Pending patent applications may never proceed to be granted patents. It is not generally possible to commence a patent
enforcement action based on a pending application; it is necessary to obtain a granted patent. However, damages in
some instances and in some jurisdictions may be backdated for part of the period of pendency. To our knowledge, none
of the patents and patent applications in-licensed or owned by us are presently the subject of a challenge by a third-
party. EP0956506 has previously been challenged in opposition proceedings before the European Patent Office but the
opposition was successfully dismissed.
Trademarks
Registered trademarks protect indications which serve to distinguish the goods or services of one competitor from those
of others, and provide the trademark owner with the exclusive right to use or authorize others to use the trademark in
relation to the goods and services for which it is registered. Trademarks are granted generally on a national or territorial
basis. International filings are governed by international treaties to simplify the process of securing trademark protection
at the national or territorial level of parties to the relevant international treaties, in a similar manner to patents. In
particular, international trademark applications made through the World Intellectual Property Organization under the
Madrid Protocol offer a six-month priority period from the application filing date. Generally, the intellectual property
offices in each country or territory conduct searches and examination of trademark applications prior to registration.
Trademark applications are subject to the pace of examination in the relevant national intellectual property office. It is
not unusual for a trademark application to be pending for a period of six months to two years prior to grant, but some
trademark applications may be pending for years. Trademarks may be subject to challenge by third parties in most
jurisdictions before and after registration, with many national or territorial intellectual property offices offering
administrative and/or judicial processes to address challenges to the relevant trademark on various grounds, which often
include likelihood of confusion concerns with senior trademark registrations.
In total, as of December 31, 2025, we own 26 registered U.S. trademarks, 14 pending U.S. trademark applications, 179
foreign trademarks registered in jurisdictions such as Australia, Europe, China, Brazil and Japan, and 93 pending foreign
trademark applications applied for in jurisdictions such as Australia, Europe, China, Brazil and Japan. We currently have
trademark registrations in the U.S. for the Telix Pharmaceuticals name, the Illuccix name and logo, the Gozellix name and
logo, the Pixclara name and logo, the ANMI name, the SENSEI name, the ARTMS name, and the RADMAB name and other
trademarks are pending in the U.S. such as Zircaix. Outside of the U.S., Illuccix is registered in Australia, Brazil, Canada,
China, the European Union, India, Israel, Japan, Malaysia, New Zealand, Norway, Peru, Philippines, South Korea,
Singapore, Switzerland, Taiwan, Türkiye, the United Kingdom and is pending in Thailand. We have also selectively filed
the following names and logos outside of the U.S.: Pixclara, Gozellix, Zircaix, ANMI, ARTMS, and RADMAB.
Data and Market Exclusivity Provisions
Data and market exclusivity provisions relating to the regulatory approval of pharmaceutical products exist in each
relevant jurisdiction. The provisions provide periods during which a competitor is limited in their ability to seek and/or
obtain regulatory approval for a generic or similar product. Data exclusivity relates to the period in which information
relating to the safety and efficacy of a product, provided to a regulatory authority for the purposes of obtaining
regulatory approval, remains confidential, or cannot be relied upon by the regulatory authority or a third-party in order to
obtain regulatory approval of another. Data exclusivity is separate from other forms of exclusivity, such as the monopoly
provided by patents. In some instances, the period of data exclusivity may extend beyond the term of any patent which
protects the same product. Market exclusivity refers to a period where a party wishing to sell a product is prohibited
from doing so, even if regulatory approval has been obtained.
As our key products are radio pharmaceutical products, they will have the benefit of periods of data and market
exclusivity available in each jurisdiction following regulatory approval. These are typically five years or more in duration
(and eight years data exclusivity plus two years market exclusivity for European jurisdictions).
Our Patent Portfolio
Our commercial success depends in part on our ability to obtain and maintain regulatory exclusivity, proprietary or
intellectual property protection for our products and product candidates, our core technologies, and other know-how, to
operate without infringing on the proprietary rights of others and to prevent others from infringing our proprietary or
intellectual property rights. Our policy is to seek to protect our proprietary and intellectual property position by, among
other methods, filing patent applications in the U.S. and in foreign jurisdictions related to our proprietary technology and
products and product candidates. We also rely on trade secrets, know-how and continuing technological innovation to
develop and maintain our proprietary and intellectual property position.
118
We also in-license patent portfolios relating to our product pipeline and to emerging product candidates as well as
technologies that are adjacent such as radiolabeling technologies, linker technologies, chelator technologies,
bioconjugation techniques, antibody manufacturing and modifications, isotope manufacture, AI techniques and
applications, and medical imaging devices,
In total, as of December 31, 2025, we have in-licensed 40 U.S.-issued patents and 408 foreign-issued patents granted in
jurisdictions such as Australia, Canada, Germany, Italy, Spain, the United Kingdom, France, Türkiye, Russia, Japan, China,
Korea, Singapore, India, Israel, Mexico, and Brazil. As of December 31, 2025, we have also in-licensed 10 pending non-
provisional U.S. patent applications, 66 pending foreign-patent applications applied for in jurisdictions such as in
Australia, Canada, Europe, Russia, Japan, China, India, Mexico, and Brazil, and one pending international application filed
under the PCT. The PCT is an international patent law treaty that provides a unified procedure for filing a single initial
patent application to seek patent protection for an invention simultaneously in each of the member states. Although a
PCT application is not itself examined and cannot issue as a patent, it allows the applicant to seek protection in any of
the member states through national-phase applications.
In total, as of December 31, 2025, we own either solely, or jointly with our commercial partners, 15 U.S.-issued patents
and 118 foreign patents granted in jurisdictions such as Australia, Canada, Germany, Italy, Spain, the United Kingdom,
France, Türkiye, Russia, Japan, China, India, Israel, Mexico, and Brazil. As of December 31, 2025, we also have pending,
either solely or jointly with our commercial partners, 38 non-provisional U.S. patent applications, 182 foreign patent
applications applied for in jurisdictions such as in Australia, Canada, Europe, Japan, China, Korea, Singapore, India, Israel,
Mexico, and Brazil, and 18 pending international applications filed under the PCT.
The intellectual property portfolios for our key products and product candidates as of December 31, 2025 are
summarized below:
Illuccix and Gozellix
Our Illuccix patent portfolio covers the pharmaceutical product and the unique arrangement of components of the kit as
well as methods of making gozetotide. The patent family directed to the pharmaceutical product and the unique
arrangement of components of the kit consists of five U.S.-issued patents; 53 foreign-issued patents granted in
Australia, Canada, Belgium, Finland, Switzerland, Lichtenstein, the Czech Republic, Denmark, Austria, Greece, Hungary,
Ireland, the Netherlands, Norway, Portugal, Sweden, Germany, Italy, Spain, the United Kingdom, France, Türkiye, Russia,
Japan, China, India, Israel, Mexico, South Africa, New Zealand and Brazil, six pending foreign patent applications applied
for in Europe, India and Hong Kong, and four pending U.S. non-provisional applications. The patent family directed to
methods of making gozetotide consists of one pending U.S. non-provisional patent application and ten pending foreign
patent applications in Australia, Brazil, Canada, Mexico, China, Europe, Japan, Korea, Hong Kong and Singapore.
There is one U.S. patent registered under the U.S. Orange Book which is directed to methods of imaging using the
pharmaceutical product prepared with Illuccix.
Any patents that may issue in the U.S. as part of our patent portfolio directed to the pharmaceutical product or the kit will
expire no earlier than 2035, not including any terminal disclaimer, patent term adjustment due to administrative delays by
the U.S. Patent and Trademark Office ("USPTO") or patent term extension under the Hatch-Waxman Act. Any patents
that may issue in foreign jurisdictions will likewise expire no earlier than 2035. Any patents that may issue in the U.S.
directed to methods of making gozetotide will expire in 2042, absent any terminal disclaimer, patent term adjustment
due to administrative delays by the USPTO or patent term extension under the Hatch-Waxman Act. Any patents issued in
foreign jurisdictions will likewise expire in 2042.
The Illuccix patent portfolio also covers Gozellix. In addition, there is one patent family directed solely to the formulation
of Gozellix comprising one pending U.S. non-provisional application and one PCT application which has yet to enter the
National Phase. Any patents that may issue in the U.S. as part of our patent portfolio directed to the pharmaceutical
product or the kit will expire no earlier than 2045, not including any terminal disclaimer, patent term adjustment due to
administrative delays by the USPTO, or patent term extension under the Hatch-Waxman Act. Any patents that may issue
in foreign jurisdictions will likewise expire no earlier than 2045.
TLX250-Px (89Zr-DFO-girentuximab) and TLX250-Tx (177Lu-DOTA-girentuximab)
We have in-licensed six patent families from Heidelberg Pharma AG (formerly Wilex AG) directed to the CAIX-targeting
girentuximab antibody and various therapeutic and imaging applications thereof.
The in-licensed patent portfolio includes three U.S.-issued patents, 17 foreign-issued patents granted in Australia,
Canada, Germany, Spain, Italy, France, the United Kingdom, Korea, New Zealand, South Africa, Israel, Russia and Mexico,
and one foreign patent applications applied for in China. Expiry dates vary from 2026 to 2034 across the portfolio, not
including any patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984,
commonly referred to as the Hatch-Waxman Act, or equivalent provisions in foreign jurisdictions.
We have two patent families directed to aspects of the manufacture of TLX250-Px. These patent families include two
pending U.S. non-provisional patent applications and 16 foreign patent applications in Australia, Brazil, Canada, Europe,
Korea, Singapore, China and Japan. Any patents that may issue in the U.S. based on the non-provisional U.S. patent
applications will expire no earlier than 2042, not including any terminal disclaimer, patent term adjustment due to
119
administrative delays by the USPTO or patent term extension under the Hatch-Waxman Act. Any patents issued in
foreign jurisdictions will likewise expire no earlier than 2042.
We have one patent family directed to the use of TLX250-Px in imaging CAIX-expressing cancers other than ccRCC. This
patent family consists of one pending U.S. non-provisional patent application and nine foreign patent applications in
Brazil, Singapore, China, Mexico, Australia, Canada, Japan, Korea and Europe. Any patents that may issue in the U.S.
based on the pending PCT application will expire no earlier than 2043, not including any terminal disclaimer, patent term
adjustment due to administrative delays by the USPTO or patent term extension under the Hatch-Waxman Act. Any
patents issued in foreign jurisdictions will likewise expire no earlier than 2043.
We have one patent family directed to the use of TLX250-Tx in therapy of CAIX-expressing cancers other than ccRCC.
This patent family consists of one pending PCT application. Any patents that may issue in the U.S. based on the pending
PCT application will expire no earlier than 2043, not including any terminal disclaimer, patent term adjustment due to
administrative delays by the USPTO or patent term extension under the Hatch-Waxman Act. Any patents issued in
foreign jurisdictions will likewise expire no earlier than 2043.
We have one patent family directed to combinations of TLX250-Tx with checkpoint inhibitors. This patent family consists
of one pending U.S. non-provisional patent application and 6 foreign patent applications applied for in Australia, Brazil,
Canada, Europe, Mexico and Singapore. Any patents that may issue in the U.S. based on the pending PCT application will
expire no earlier than 2043, not including any terminal disclaimer, patent term adjustment due to administrative delays by
the USPTO or patent term extension under the Hatch-Waxman Act. Any patents issued in foreign jurisdictions will
likewise expire no earlier than 2043.
We have one patent family directed to the combination of TLX250-Tx with DNA damage repair inhibitors. This patent
family includes one pending U.S. non-provisional patent application and ten foreign patent applications applied for in
Australia, Brazil, Canada, Europe, Israel, Korea, Mexico, Singapore, China and Japan, Any patents that may issue in the
U.S. based on the U.S. non-provisional patent application will expire no earlier than 2042, not including any terminal
disclaimer, patent term adjustment due to administrative delays by the USPTO or patent term extension under the
Hatch-Waxman Act. Any patents issued in foreign jurisdictions will likewise expire no earlier than 2042.
As biological products, TLX250-Px and TLX250-Tx will be entitled to 12 years data exclusivity from the date of product
approval.
TLX252-Tx (225Ac-DOTA-girentuximab)
We have a single patent family patent directed to the composition of matter of TLX252-Tx, its radiolabeled forms and
uses in imaging and therapy. The patent family includes one pending U.S. non-provisional patent application and 16
pending foreign patent applications in Canada, Chile, China, India, Japan, Korea, Mexico, Australia, Europe, Eurasia, India,
New Zealand, Brazil, Hong Kong, Israel and Singapore. Any patents that may issue in the U.S. will expire no earlier than
2040, not including any terminal disclaimer, patent term adjustment due to administrative delays by the USPTO or patent
term extension under the Hatch-Waxman Act. Any patents that may issue in foreign jurisdictions will likewise expire no
earlier than 2040.
TLX101-Px (18F-FET)
We have orphan drug and fast track designation for TLX101-Px in the U.S., which we expect to yield up to seven years
regulatory exclusivity following product approval.
TLX101-Tx (131I-IPA) and TLX102-Tx (211At-APA)
We have in-licensed a patent portfolio directed to methods of treatment using TLX101-Tx and TLX102-Tx from Dr.
Samuel Samnick, a German nuclear medicine researcher. There are two U.S.-issued patents which will expire no earlier
than 2028 and 2031 respectively, not including any patent term extension under the Hatch-Waxman Act. There are eight
foreign issued patents in Australia, Canada, Germany, the United Kingdom, Spain, France, Japan, and Korea which will
expire no earlier than 2026.
We have in-licensed a patent portfolio directed to a method of manufacturing TLX101-Tx and TLX102-Tx from Osaka
University. There are two U.S.-issued patents, one pending U.S. non-provisional application, seven foreign-issued
patents in Australia, Japan, Switzerland, Lichtenstein, Spain, the United Kingdom, and Türkiye, one foreign issued patent
under the European Unified Patent Convention (which covers Austria, Belgium, Bulgaria, Denmark, Estonia, Finland,
France, Germany, Italy, Latvia, Lithuania, Luxembourg, Malta, Netherlands, Portugal, Romania, Slovenia, and Sweden) and
two pending foreign applications in Europe and Hong Kong. Any patents in this portfolio that may issue in the U.S. will
expire no earlier than 2038, not including any terminal disclaimer, patent term adjustment due to administrative delays by
the USPTO or patent term extension under the Hatch-Waxman Act. Any patents issued in foreign jurisdictions will
likewise expire no earlier than 2038.
We have one PCT application directed to combinations of TLX101-Tx and other chemotherapeutic agents. Any patents
that may issue in the U.S. based on this PCT application will expire no earlier than 2045, not including any terminal
disclaimer, patent term adjustment due to administrative delays by the USPTO or patent term extension under the
Hatch-Waxman Act. Any patents issued in foreign jurisdictions will likewise expire no earlier than 2045.
120
We have orphan drug designation for TLX101-Tx in the U.S. and Europe and for TLX102-Tx in the U.S. which will grant us
the customary regulatory exclusivity, currently expected to be up to seven years from date of product approval.
TLX591-Tx (lutetium (Lu177) rosopatamab tetraxetan)
We have sub-licensed a Cornell University (and associated entities) patent portfolio from Convergent Therapeutics, Inc
as successor in title of BZL Biologics LLC. The portfolio is directed to TLX591-Tx and combination therapies of TLX591-
Tx with androgen deprivation therapy.
The sub-licensed patent portfolio includes one U.S.-issued patent, one pending U.S. non-provisional patent application,
14 foreign-issued patents in Belgium, Canada, Japan, Germany, France, Spain, the United Kingdom, Luxembourg and the
Netherlands, and a pending foreign application in Europe directed to combinations with androgen deprivation therapy.
Any patents that may issue in the U.S. will expire no earlier than 2028, not including any terminal disclaimer, patent term
adjustment due to administrative delays by the USPTO or patent term extension under the Hatch-Waxman Act. Any
patents that may issue in foreign jurisdictions will likewise expire no earlier than 2028.
As a biological product, TLX591-Tx will be entitled to 12 years data exclusivity from the date of product approval.
TLX592-Tx (225Ac-RADmAb®)
We have a single patent family patent directed to the composition of matter of TLX592-Tx, its radiolabeled forms and
uses in imaging and therapy. The patent family includes one pending U.S. non-provisional patent application, one granted
Japanese patent, and 16 pending foreign patent applications in Canada, Chile, China, India, Japan, Korea, Mexico,
Australia, Europe, Eurasia, India, New Zealand, Brazil, Hong Kong, Israel and Singapore. Any patents that may issue in the
U.S. will expire no earlier than 2040, not including any terminal disclaimer, patent term adjustment due to administrative
delays by the USPTO or patent term extension under the Hatch-Waxman Act. Any patents that may issue in foreign
jurisdictions will likewise expire no earlier than 2040.
TLX592-Tx is currently in early development so regulatory pathway to product approval is not yet confirmed or known,
however the customary regulatory exclusivity period is expected to apply.
TLX300-Px (89Zr-olaratumab) and TLX300-Tx
We have two patent families directed to radiolabeled forms of olaratumab and their use in imaging and therapy. The
patent family includes two pending U.S. non-provisional patent applications and 16 pending foreign applications filed in
Australia, Brazil, Canada, China, Europe, Japan, Korea, and Mexico. Any patents that may issue in the U.S. will expire no
earlier than 2043, not including any terminal disclaimer, patent term adjustment due to administrative delays by the
USPTO or patent term extension under the Hatch-Waxman Act. Any patents that may issue in foreign jurisdictions will
likewise expire no earlier than 2043.
TLX66-Px (99mTc-besilesomab, Scintimun) and TLX66-Tx (90Y-DTPA-besilesomab)
We have one patent family directed to the use of TLX66-Tx in the treatment of multiple myeloma. The patent family
includes one U.S.-issued patent and foreign-issued patents in Canada, Australia, and Europe (validated in Belgium,
Germany, Spain, France, the United Kingdom and Italy). The U.S. patent has a maximum expiry date of 2031, not
including any patent term extension under the Hatch-Waxman Act. The other patents will expire no earlier than 2026.
A second patent family is directed to the use of TLX66-Tx for treating AL-amyloidosis and for specific bone-marrow
conditioning. We have one pending U.S. non-provisional patent application and pending applications in China, Japan and
Canada.
The second family includes a granted patent in Europe which has been validated under the unitary patent system (which
covers seventeen European countries and includes coverage of Belgium, Germany, France, and Italy) and has also been
validated in Switzerland, Spain, and the United Kingdom. There are also foreign-issued patents in Australia and South
Africa.
Any patents in the U.S. that may issue in the second family will expire no earlier than 2038, not including any terminal
disclaimer, patent term adjustment, or patent term extensions under the Hatch-Waxman Act. Any patents issued in
foreign jurisdictions will likewise expire no earlier than 2038.
We have one PCT application directed to the use of TLX66-Px (Scintimun) in dosimetry. Any patents that may issue in
the U.S. based on the pending PCT application will expire no earlier than 2045, not including any terminal disclaimer,
patent term adjustment due to administrative delays by the USPTO or patent term extension under the Hatch-Waxman
Act. Any patents issued in foreign jurisdictions will likewise expire no earlier than 2045.
TLX090-Tx (153Sm-DOTMP)
We have in-licensed three patent families from IGL Pharma, Inc in connection with our acquisition of QSAM Biosciences,
Inc., which are directed to methods of manufacturing TLX090-Tx, kits comprising TLX090-Tx, and its use in treatment.
121
The portfolio includes five U.S.-issued patents, 39 foreign-issued patents in Canada, Germany, France, Great Britain,
Hungary, Ireland, Iceland, Italy, Luxembourg, the Netherlands, Norway, Austria, Belgium, Bulgaria, the Czech Republic,
Denmark, Finland, Poland, Portugal, Slovakia, Slovenia, Sweden, Switzerland, Türkiye and Japan, and three pending
foreign patent applications in Japan and Europe. Any patents issued in the U.S. will expire no earlier than 2035, not
including any terminal disclaimer or patent term adjustment due to administrative delays by the USPTO or patent term
extension under the Hatch-Waxman Act. Any patents issued in foreign jurisdictions will likewise expire no earlier than
2035.
TLX400-Tx (177Lu-DOTAGA.Glu.(FAPi)2)
We have one patent family directed to the composition of matter of TLX400-Tx. The family includes one granted German
utility patent, one pending U.S. non-provisional patent application and nine foreign patent applications in Australia, Brazil,
Canada, China, Europe, India, Japan, Korea and Hong Kong. The granted German utility patent will expire no earlier than
2031. Any patents issued or that may issue based on the pending application in the U.S. will expire no earlier than 2042,
not including any terminal disclaimer or patent term adjustment due to administrative delays by the USPTO or patent
term extension under the Hatch-Waxman Act. Any patents issued in foreign jurisdictions will likewise expire no earlier
than 2042.
TLX593-Px
We have in-licensed a PCT application from the University of Ghent directed to methods of preparing TLX593-Px and
associated compositions of matter. Any patents issued or that may issue based on the pending PCT application in the
U.S. will expire no earlier than 2045, not including any terminal disclaimer or patent term adjustment due to
administrative delays by the USPTO or patent term extension under the Hatch-Waxman Act. Any patents issued in
foreign jurisdictions will likewise expire no earlier than 2045.
TLX597-Tx
We have in-licensed a patent family directed to the composition of matter of TLX597-Tx from Instituto Nacional de
Investigaciones Nucleares ("ININ") a nuclear medicine research organization. The license covers all territories except
Mexico and South Africa. The patent family includes one U.S. issued patent and foreign issued patents in Eurasia, China,
Hong Kong, the United Kingdom, Switzerland and Spain, and one foreign issued patent under the European Unified
Patent Convention (which covers Austria, Belgium, Bulgaria, Denmark, Estonia, Finland, France, Germany, Italy, Latvia,
Lithuania, Luxembourg, Malta, Netherlands, Portugal, Romania, Slovenia, and Sweden). There are three foreign patent
applications filed in Brazil, Canada, and Egypt. Any patents issued or that may issue in the U.S. will expire no earlier than
2039, not including any terminal disclaimer or patent term adjustment due to administrative delays by the USPTO or
patent term extension under the Hatch-Waxman Act. Any patents issued in foreign jurisdictions will likewise expire no
earlier than 2039.
We have one unpublished PCT application directed to novel radiolabeled forms of TLX597-Tx. Any patents that may
issue in the U.S. based on the pending PCT application will expire no earlier than 2045, not including any terminal
disclaimer, patent term adjustment due to administrative delays by the USPTO or patent term extension under the
Hatch-Waxman Act. Any patents issued in foreign jurisdictions will likewise expire no earlier than 2045.
RHN001-Tx and RHN001-Dx
We have one patent family directed to RHN001-Tx and RHN001-Dx as a composition of matter. The patent family
includes one U.S non-provisional patent application and patent applications in Europe, Canada, Australia, India, South
Africa, Mexico, China, Eurasia, Brazil and Hong Kong. Any patents that may issue in the U.S. will expire no earlier than
2042, not including any terminal disclaimer, patent term adjustment due to administrative delays by the USPTO or patent
term extension under the Hatch-Waxman Act. Any patents issued in foreign jurisdictions will likewise expire no earlier
than 2042.
TLR602-Tx and TLR603-Tx
We have one PCT application directed to TLX602-Tx as a composition of matter. Any patents that may issue in the U.S.
based on the pending PCT application will expire no earlier than 2044, not including any terminal disclaimer or patent
term adjustment due to administrative delays by the USPTO or patent term extension under the Hatch-Waxman Act. Any
patents issued in foreign jurisdictions will likewise expire no earlier than 2044.
We have one patent family directed to TLX603-Tx as a composition of matter. The patent family includes one U.S non-
provisional patent application and patent applications in Europe, China, Korea, Mexico, Japan, Singapore, Brazil,
Australia, Canada, and Hong Kong. Any patents that may issue in the U.S. based on the pending PCT application will
expire no earlier than 2043, not including any terminal disclaimer, patent term adjustment due to administrative delays by
the USPTO or patent term extension under the Hatch-Waxman Act. Any patents issued in foreign jurisdictions will
likewise expire no earlier than 2043.
Lightpoint Medical
In connection with our acquisition of Lightpoint’s radio-guided surgery business, we acquired a patent portfolio relating
to surgical applications of radiopharmaceuticals including the SENSEI probe. The patent portfolio comprises five U.S.-
122
issued patents, 25 foreign-issued patents in Australia, China, Belgium, Luxembourg, Switzerland, Germany, Italy, France,
the Netherlands, Spain and the United Kingdom, one granted European patent which has been validated in Great Britain,
Spain and under the European Unified Patent Convention (which covers Austria, Belgium, Bulgaria, Denmark, Estonia,
Finland, France, Germany, Italy, Latvia, Lithuania, Luxembourg, Malta, Netherlands, Portugal, Romania, Slovenia, and
Sweden), and two pending non-provisional U.S. applications. Any patents issued or that may issue based on the pending
applications in the U.S. will expire no earlier than 2033, not including any terminal disclaimer or patent term adjustment
due to administrative delays by the USPTO or patent term extension under the Hatch-Waxman Act. Any patents issued in
foreign jurisdictions will likewise expire no earlier than 2033.
Collaboration and License Agreements
Advanced Nuclear Medicine Ingredients SA
In December 2018, we acquired Advanced Nuclear Medicine Ingredients ("ANMI") including the pre-cursor kit that was
ultimately developed to become Illuccix. We paid A$2.7 million in cash and issued 6,090,805 ordinary shares, based on a
share price of A$0.637 per share, in connection with the closing of the acquisition.
We are obligated to make deferred earn-out payments to former shareholders of ANMI on an annual basis equal to a
percentage in the low teens of net sales of Illuccix in the U.S. and equal to a percentage in the low twenties of net sales
of Illuccix outside the U.S., in each case until April 13, 2027, which is five years following the first commercial sale of
Illuccix in the U.S. We hold an option to buy out the remaining deferred payments by paying €10 million within 90 days of
April 13, 2025. This option was exercised and the final buy out payment was made in July 2025.
Heidelberg License Agreement
On January 16, 2017, we entered into a license agreement, or, as amended, the Heidelberg License, with Wilex AG (now
Heidelberg Pharma AG, "Heidelberg"), pursuant to which Heidelberg granted us an exclusive, royalty-bearing license
under certain patents and know-how to develop, manufacture and commercialize the CAIX-targeting girentuximab
antibody ("girentuximab"), radio-labeled with an isotope in both diagnostic and therapeutic products. We paid Heidelberg
US$250,000 in connection with the execution of the Heidelberg License and initial technology transfer. In addition, from
2018 to 2022, we have paid Heidelberg US$1.25 million for achievement of certain manufacturing and regulatory
milestones for IND approval and enrollment of the last patient in a Phase 3 clinical trial. Under the agreement, we are
obligated to pay milestone payments to Heidelberg of US$2.4 million in the aggregate with payment owed upon FDA
approval for a BLA for a diagnostic product and upon first reimbursements for first indication of a diagnostic product.
Under the Heidelberg License, Heidelberg retained the right to develop and commercialize products that contain
girentuximab that are not radio-labeled. In the event we intend to file a BLA for a therapeutic product that includes
girentuximab, we are obligated to notify Heidelberg and may be required to pay up to US$3.0 million to extinguish any of
Heidelberg’s retained rights that have been granted to a third-party for co-promotion of girentuximab in the U.S. In the
event of commercial launch of a diagnostic product, we are obligated to pay Heidelberg royalties in the low twenties on
net sales of such product by us or a sublicensee during the first ten years of such sales and mid single-digit royalties on
net sales of such product during the second ten years of such sales. In the event of commercial launch of a therapeutic
product, we are obligated to pay Heidelberg low single-digit royalties on net sales of such product during the first ten
years of such sales. Our obligation to pay royalties on net sales of diagnostic products expires 20 years after first
commercial sale of each diagnostic product and our obligation to pay royalties on therapeutic products expires ten years
after first commercial sale of each therapeutic product. We are obligated to use commercially reasonable efforts to
develop products for regulatory approval worldwide subject to certain excepted countries for therapeutic products. The
Heidelberg License expires when we cease selling products subject to the license granted thereunder, subject to
customary termination provisions regarding material breach by or bankruptcy of either party. In addition, we can
terminate the agreement upon 180 days’ written notice for any reason. In the event of termination of the agreement for
Heidelberg’s material breach or bankruptcy, we have the option to purchase intellectual property relating to the products
for nominal consideration.
On March 1, 2024, Heidelberg assigned its rights and obligations under the Heidelberg License to HDP G250, AG & Co.
KG, a wholly owned subsidiary of Heidelberg. In connection with the assignment, the subsidiary agreed to perform all
obligations of Heidelberg under the Heidelberg License. On March 4, 2024, Heidelberg announced that it entered into a
royalty financing agreement with HealthCare Royalty Partners relating to royalty payments that Heidelberg is entitled to
receive from us under the Heidelberg License.
Olaratumab License Agreement
In April 2022, we entered into a license agreement ("the Lilly License") with Eli Lilly Kinsale Limited, pursuant to which
Lilly granted us an exclusive, royalty-bearing license under certain patents and know-how directed to its proprietary
antibody, olaratumab, to develop, manufacture and commercialize radio-labeled forms of olaratumab for the diagnosis
and treatment of human cancers. Under the Lilly License, we are obligated to use commercially reasonable efforts to
develop, obtain regulatory approval for and commercialize radio-labeled forms of olaratumab in several major markets.
As consideration for the Lilly License, we paid Lilly an upfront payment of US$5.0 million and are obligated to pay up to a
total of US$225.0 million upon satisfaction of specified clinical, regulatory and commercial milestones. In the event of
launch of a commercial product, we are also obligated to pay Lilly royalties in the low teens based on net sales of
products, with the royalty term being, on a product-by-product and country-by-country basis, the latest of (i) the 12th
anniversary of the first commercial sale of such product in such country, (ii) the first day on which there is not at least
one of Lilly’s patents covering such product in such country, or (iii) the expiration of the last-to-expire data exclusivity
123
period for such product in such country, which we refer to as the Telix Royalty Term. The royalties may also be subject to
reductions during the Telix Royalty Term in the event the product is not covered by a valid claim of a licensed patent or
in the event we are required to obtain a license from a third-party to commercialize the product. In addition to the
foregoing royalties on net sales of products, we are obligated to pay Lilly a percentage of any sublicense revenue
received pursuant to any sublicense or similar agreement. The Lilly License defines sublicense revenue to include
amounts paid for milestones similar to the milestones specified in the Lilly License, but solely to the extent such amounts
are above the amount paid to Lilly under the Lilly License, and further defines sublicense revenue to exclude royalties
calculated on the basis of sales of the product for which royalties are already due under the Lilly License, reimbursement
for patent costs, certain profit sharing payments and any equity or debt investment at fair market value. The Lilly License
further specifies that the royalty owed on such sublicense revenue varies based on the date we enter into such
sublicense or similar agreement, ranging from mid-teens if entered into within one (1) year of the effective date of the
Lilly License down to mid-single digits if entered into following the third (3rd) anniversary of the effective date of the Lilly
License.
Under the Lilly License, we also granted Lilly an option to enter into an exclusive license under certain patents and know-
how to develop, commercialize and otherwise exploit a companion diagnostic for use with olaratumab, or the companion
diagnostic option. To exercise the companion diagnostic option, Lilly is obligated to pay us an option exercise fee of
US$5.0 million and would be obligated to pay up to a total of US$30.0 million upon satisfaction of specified regulatory
milestones. In the event of launch of a companion diagnostic, Lilly would pay us low single digit royalties on net sales of
its products incorporating olaratumab, or Lilly Products, for the labeled use for the treatment of human cancer and mid-
single digit royalties on net sales of companion diagnostics, with the royalty term being, on a product-by-product and
country-by-country basis, the latest of (i) the 12th anniversary of the first commercial sale of such Lilly Product or
companion diagnostic in such country, (ii) the first day on which there is not at least one of our patents covering such
Lilly Product or companion diagnostic in such country, or (iii) the expiration of the last-to-expire data exclusivity period
for such Lilly Product or companion diagnostic in such country, or the Lilly Royalty Term. The royalties may also be
subject to reductions during the Lilly Royalty Term in the event the Lilly Product or companion diagnostic is not covered
by a valid claim of a licensed patent or in the event Lilly is required to obtain a license from a third-party to commercialize
the product.
The Lilly License continues until the expiration of the last-to-expire Telix Royalty Term or, if Lilly exercises the companion
diagnostic option, the Lilly Royalty Term, subject to customary termination provisions regarding material breach by or
bankruptcy of either party. Each party, in its capacity as the licensee, may terminate the agreement with respect to the
licenses granted to it upon 30 days’ written notice to the other party. Lilly may terminate the agreement if a patient has
not been enrolled in a Phase 1 or Phase 2 clinical trial using the companion diagnostic by April 8, 2025. If Lilly exercises
the companion diagnostic option, we may terminate the agreement if no patient has qualified for enrollment into a
registrational study of the Lilly Product for use by patients that have been screened using the companion diagnostic
within two years of the date that Lilly exercised the companion diagnostic option.
Lightpoint Medical Share Sale Agreement
In June 2023, we entered into a share sale agreement to acquire the SENSEI business from Lightpoint. We completed the
acquisition of Lightpoint on November 1, 2023. The acquisition was implemented through the purchase of Lightpoint
Medical Limited’s wholly owned subsidiary, Lightpoint Surgical Limited, as the then owner of Lightpoint’s business, assets
and operation. We paid upfront consideration of US$20.0 million, of which we paid US$19.6 million through the issuance
of 3,298,073 ordinary shares at a price of A$9.3659 per share. We are obligated to pay an additional US$15.0 million via
an earn-out in the form of performance rights, which may be settled in cash or ordinary shares, at our option, upon
achievement of regulatory, commercial and operational milestones relating to the ongoing development and
commercialization of SENSEI. As at 31 December 2025, US$4.5 million of these performance rights have been settled by
the issue of 269,075 ordinary shares.
Strategic License and Commercial Partnership with Grand Pharma
In November 2020, we entered into a strategic partnership with Grand Pharma, pursuant to which we appointed Grand
Pharma as our partner with exclusive development and commercialization rights to our portfolio of imaging and
therapeutic products and product candidates in Mainland China, Taiwan, Hong Kong and Macau ("the Grand Pharma
Territory"). As part of the strategic partnership, we entered into an Imaging Products Commercialization Agreement and a
Therapeutic Products License Agreement with Grand Pharma.
Pursuant to the Imaging Products Commercialization Agreement, we appointed Grand Pharma as our exclusive
commercial partner in the Grand Pharma Territory for Illuccix and TLX250-Px. The Imaging Products Commercialization
Agreement includes minimum annual purchase obligations of Grand Pharma following marketing authorization in
applicable regions in the Grand Pharma Territory in order to maintain exclusivity in the Grand Pharma Territory.
There are currently no approved imaging products in the Grand Pharma Territory under the Imaging Products
Commercialization Agreement.
The Imaging Products Commercialization Agreement has a 15-year term for each product beginning on the date of
marketing authorization in China and the agreement will automatically renew for five-year renewal terms unless either
party gives a written notice of nonrenewal. Either party may terminate the Imaging Products Commercialization
Agreement upon material breach or insolvency by the other party.
124
Pursuant to the Therapeutic Products License Agreement, Grand Pharma is responsible, at its own cost, for conducting
any clinical trials of therapeutic products in the Grand Pharma Territory in accordance with the agreed development plan.
Pursuant to the Therapeutic Products License Agreement, we are eligible to receive payments of up to US$69.0 million
upon achievement of regulatory milestones with respect to therapeutic products by Grand Pharma and up to US$156.0
million upon achievement of commercial milestones with respect to therapeutic products by Grand Pharma. We are also
eligible to receive single-digit percentage royalties on net sales of therapeutics products in the Grand Pharma Territory
for ten years after marketing authorization is granted in the Grand Pharma Territory. There are currently no approved
therapeutic products in the Grand Pharma Territory and we have not received any milestone payments from Grand
Pharma under the Therapeutic Products License Agreement.
We received an upfront, non-refundable cash payment of US$25.0 million upon execution of the Therapeutic Products
License Agreement. This upfront payment will be credited against any regulatory or commercial milestone payments
owed to us by Grand Pharma.
The Therapeutic Products License Agreement has a ten-year term ending after the date that marketing authorization is
granted in respect of each product. Either party may terminate the Therapeutic Products License Agreement upon
material breach or insolvency by the other party.
Agreement and Plan of Merger with IsoTherapeutics Group, LLC
On February 27, 2024, we entered into an agreement and plan of merger ("the IsoTherapeutics Agreement") to acquire
IsoTherapeutics Group, LLC ("IsoTherapeutics"), a specialty radiopharmaceutical development and bioconjugation firm,
based in Texas. IsoTherapeutics provides radiochemistry and bioconjugation development and contract manufacturing
services to many companies in the radiopharmaceutical industry. We completed the acquisition of IsoTherapeutics on
April 9, 2024.
The acquisition has enhanced our internal drug development capabilities, enabling us to internalize select aspects of our
development programs, with the goal of reducing cost and time to achieve technical milestones. The acquisition
expanded our U.S. manufacturing footprint with a site that includes a GMP clean room and production infrastructure
suitable for clinical use. The site also has extensive capacity to process a wide variety of therapeutic isotopes used in our
development portfolio. We are realizing cost savings from internalizing radiochemistry-related R&D activities, as
demonstrated by the inter-segment activity during the year.
The purchase price for the acquisition consists of (i) US$8.1 million paid at closing in the form of US$2.1 million in cash
and US$6.0 million in our ordinary shares which we paid in the form of 717,587 of our ordinary shares issued at closing,
(ii) US$5.0 million in performance-related milestone payments, which are payable in cash and are subject to meeting
certain milestone conditions within 12 months of closing, and (iii) a two-year revenue share that is based on actual
revenue earned from existing customers of IsoTherapeutics, which we estimate will require total cash payments of
approximately US$0.6 million. The upfront cash consideration was subject to customary working capital, debt and
transaction expense adjustments. The number of shares issued at closing was determined by converting US$6.0 million
to Australian dollars using the Reserve Bank of Australia exchange rate at closing and dividing that amount by the volume
weighted average price at which our ordinary shares traded on the ASX over the 10-trading day period prior to closing.
The shares issued at closing are subject to voluntary escrow restrictions.
The US$5.0 million in performance-related milestones was paid during the year following satisfaction of the performance
conditions. In addition, the first year of the revenue share has been paid totalling US$0.4 million.
Share Purchase Agreement with ARTMS Inc.
On March 5, 2024, we entered into a share purchase agreement ("the ARTMS Agreement") to acquire ARTMS Inc.
("ARTMS"), a radioisotope production technology company based in Canada, and its advanced cyclotron-based isotope
production platform, manufacturing plant and stockpile of ultra-pure rare metals required for consumable target
production. We completed the acquisition of ARTMS on April 11, 2024. ARTMS is a commercial-stage company that
specializes in the physics, chemistry and materials science of cyclotron-produced radionuclides and its technology is
used by major manufacturing networks to optimize production of a range of medical radioisotopes. We expect that the
acquisition will further enhance the vertical integration of our supply chain and manufacturing by providing a greater level
of control and security over each of our diagnostic isotopes.
ARTMS’ core technology platform is based on the QUANTM Irradiation System ("QIS"), a complete cyclotron-based
isotope production system that is designed to support high efficiency and cost-effective production of commercially
important medical isotopes including zirconium-89, gallium-68, technetium-99m and copper-64. We also expect that its
advanced cyclotron technologies will have immediate application and differentiation in the production of future
commercially important alpha-emitting, therapeutic isotopes, including actinium-225 and astatine-211.
We believe that QIS may be able to produce zirconium-89 that is ready for radiopharmaceutical use with TLX250-Px by
irradiating yttrium-89. ARTMS also holds a stockpile of zinc-68, which is used to produce gallium-68 that could be used
with Illuccix. Following closing of the acquisition, we intend to work with pharmacy networks and partners to enhance the
reliability and routine production of commercially useful cyclotron-produced diagnostic radionuclides such as copper-64
and technetium-99. In particular, ARTMS has a stockpile of nickel-65, an essential raw material for copper-64
production, and which is in limited global supply. As part of the acquisition, we also acquired ARTMS’ production facility
125
and clean rooms, located in Burnaby, British Columbia. We plan to continue to operate and expand ARTMS’ R&D and
production capabilities at the Burnaby location to support our in-house and customer needs, subject to applicable laws
and transaction terms.
The purchase price for the acquisition consists of: (i) US$57.5 million upfront consideration, US$15.0 million of which we
paid in cash and the balance of which we paid in the form of 5,674,635 of our ordinary shares issued at closing, (ii)
US$24.5 million million in contingent future earn out payments, payable in cash following achievement of certain
regulatory and commercial milestones, and (iii) cash earnouts representing low teens percentage royalties based on net
sales of ARTMS products and related services and representing low single-digit percentage royalties based on net sales
of Telix products prepared using ARTMS products for up to three years depending on the product location where the
sale occurs. All earn-out royalties which have not otherwise expired will terminate on the 10-year anniversary following
closing of the ARTMS acquisition. The cash upfront consideration was subject to customary working capital, debt and
transaction expense adjustments. The shares issued at closing were subject to voluntary escrow restrictions. As at
31 December 2025, US$2.0 million of the contingent future earn outs have been paid following achievement of the
milestones.
Agreement and Plan of Merger with QSAM Biosciences, Inc.
On February 7, 2024, we entered into an Agreement and Plan of Merger ("the QSAM Agreement") with QSAM
Biosciences, Inc. ("QSAM") and we completed the acquisition of QSAM on May 3, 2024.
QSAM is developing therapeutic radiopharmaceuticals for primary and metastatic bone cancer. Its lead product
candidate is Samarium-153-DOTMP, (153Sm-DOTMP), which is a novel kit-based bone-seeking targeted
radiopharmaceutical candidate that uses a next generation chelating agent to deliver a proprietary formulation of
Samarium-153 radioisotope. 153Sm-DOTMP, which we have designated as TLX090-Tx, has two potential applications –
pain management of bone metastases and osteosarcoma therapy, including in pediatric patients. We believe that
TLX090-Tx is highly aligned with our existing therapeutic focus areas of prostate cancer, glioma and sarcoma.
TLX090-Tx has shown evidence of safety, efficacy and future commercial utility in pre-clinical studies and early clinical
trials. We believe that it has the potential to deliver significant improvements on prior bone-seeking agents in the
treatment and management of late-stage metastatic disease. TLX090-Tx may enable the pain management of prostate
cancer bone metastases, where there remains a significant unmet patient need particularly after progression from other
forms of radionuclide and radiation therapy. We also believe that TLX090-Tx may benefit patients with metastatic lung
and breast cancer, where many patients develop brain and bone metastases, and disease management often focuses on
quality-of-life palliative care.
TLX090-Tx has also been granted orphan drug and rare pediatric disease designations by the FDA for the treatment of
osteosarcoma. The rare pediatric disease designation may enable TLX090-Tx to be brought to market more rapidly
through regulatory incentives, including eligibility for a pediatric rare disease priority review voucher that may be applied
to this or other programs. The orphan drug designation and the rare pediatric disease designation do not increase the
likelihood of marketing approval.
The total consideration, calculated based on the announced purchase price, for the acquisition consists of: (i) US$33.1
million upfront consideration, US$27.8 million of which was paid in closing consideration through the issuance of
3,671,120 ordinary shares, and the balance of which was paid in certain cash adjustments or through the issuance of
approximately 409,026 of our ordinary shares in change of control fees, transaction bonuses and holdback shares
reserved for settlement of purchase price adjustments and (ii) up to US$90.0 million in contingent future earn-out
payments, in cash and/or ordinary shares, without interest, upon the achievement of certain regulatory and commercial
milestones, at the times and subject to the terms and conditions of the contingent value rights agreement. The ordinary
shares issued upon closing are subject to voluntary escrow conditions. The ordinary shares issued as part of the upfront
purchase price were issued pursuant to an exemption from registration under the Securities Act, in reliance on Section
4(a)(2) and Regulation D thereunder, as a transaction by an issuer not involving a public offering.
Stock Purchase Agreement with RLS (USA) Inc.
On September 20, 2024, we entered into a stock purchase agreement ("the RLS Agreement") to acquire RLS, which we
closed in January 2025. The purchase price for the acquisition consists of: (i) US$230.0 million upfront consideration,
payable in cash at closing of the acquisition, which amount will be adjusted for transaction expenses, cash and cash
equivalents (net of restricted cash), debt and debt equivalents and working capital, and (ii) milestone payments of up to
US$20.0 million in the aggregate, payable in cash upon the achievement of certain commercial milestones. The purchase
price and related transaction costs incurred prior to closing were funded from existing cash reserves.
RLS is a U.S.-based radiopharmacy company distributing PET, SPECT and therapeutic radiopharmaceuticals. Its network
includes over 30 licensed radiopharmacies located in major metropolitan areas in 18 states across the U.S. The RLS
footprint includes over 100,000 square feet of licensed expansion space that we believe can be utilized to meet rapidly
growing production demand. RLS has approximately 1,500 customers and currently is one of the distributors of Illuccix in
the U.S.
The acquisition significantly expands our North American manufacturing footprint and establishes the basis of a next
generation radiometal production network. By augmenting our existing distribution network with RLS’ capabilities, we aim
126
to provide additional supply chain back-up and improve capacity to meet future demand, while broadening access for
patients across the entire U.S. market, including under-served populations. We believe the acquisition of RLS is highly
aligned with our investment strategy to strengthen our vertically integrated supply chain and manufacturing and
distribution capabilities. We expect the acquisition to provide a pathway for deploying ARTMS’ QIS technology by
enabling us to scale up the production of key isotopes and build a stable and consistent supply of PET and SPECT
diagnostic tracers, along with therapeutic radiopharmaceuticals across the U.S.
RLS will continue to service its existing customer base and will operate as part of our Manufacturing Solutions business,
which includes ARTMS, IsoTherapeutics, TMS Brussels South in Belgium, TMS North Melbourne in Australia, TMS
Sacramento and Telix Targeting Technologies ("T3") in California (U.S.), and TMS Yokohama in Japan. We expect that
RLS will become a key node in our network of U.S. manufacturing and distribution partnerships and is geographically
complementary to our manufacturing facility in Belgium.
Asset purchase and in-license agreements for FAP-targeting theranostics
On November 19, 2024, we entered into an asset purchase agreement with Medianezia GmbH, a company based in
Wiesbaden, Germany, and a separate exclusive, worldwide license and commercialization agreement with SCV GmbH, a
company based in Berlin, Germany, which we refer to collectively as the Medianezia/SCV Transaction. The asset
purchase completed on March 12, 2025. The technology to be acquired and licensed in the Medianezia/SCV Transaction
is comprised of FAP-targeting small molecules, patents and patent applications covering such molecules and their uses,
and the related know-how for potential use in diagnostic and therapeutic applications relating to cancer and other
diseases. FAP is a cell surface protein expressed in the tumor micro-environment of epithelial cancers and on the surface
of some specific cancer types, making FAP an attractive pan-cancer marker for therapeutic and diagnostic
radiopharmaceutical applications.
Pursuant to the Medianezia/SCV Transaction, we paid a total of €0.7 million in cash in connection to the signing of the
agreements with a further €6.3 million due at closing and will pay a further €3.0 million 12 months from closing, subject
to any potential indemnity setoff. We are also obligated to pay up to €132.0 million in contingent future earn-out
payments upon achievement of certain clinical development and regulatory milestones related to both the diagnostic and
therapeutic products under both agreements and up to €20.0 million in contingent future earn-out payments upon
achievement of certain commercial milestones related to the diagnostic product under the license agreement, as well as
low- to mid- single-digit royalties on net sales of the diagnostic product and an earlier formulation of the therapeutic
product, if used.
Acquisition of ImaginAb Assets
On January 13, 2025, we entered into an asset purchase agreement with ImaginAb ("the ImaginAb Transaction") to
acquire a significant portion of the intellectual property and technology assets of ImaginAb. The acquisition closed on
January 30, 2025. Upon closing of the ImaginAb Transaction, we acquired patents, know-how and other intellectual
property comprising a proprietary drug discovery platform, a pipeline of early-stage drug candidates against high-value
targets including DLL3 and integrin αvβ6, and several other novel targets in discovery stage. The acquired intellectual
property utilizes small engineered antibody formats that enable highly specific cancer targeting, combined with fast
tumor uptake and blood clearance. We believe this technology has the potential to be highly effective for imaging and
treating tumors with a broad range of radioisotopes, with alpha emitters of particular interest.
At the closing of the ImaginAb Transaction, we also assumed the lease for a state-of-the-art research facility in
Inglewood, California, and hired members of ImaginAb’s talented team of discovery, protein engineering and
radiopharmaceutical development experts. We expect that these assets will provide us with further in-house capabilities
in antibody engineering and preclinical development, as well as a novel biologics platform to create the next generation
of Telix precision medicine and therapeutic products, beyond the current clinical-stage pipeline.
The ImaginAb Transaction was conducted through an asset purchase agreement with a concurrent technology license
agreement, the latter of which provided us additional exclusive and non-exclusive rights to certain patents and know-
how being retained by ImaginAb. The upfront purchase price for the transaction was $67,116,000 which comprised
$10,000,000 in cash and 29,895,000 in equity through the issue of 2,053,311 fully paid ordinary Telix shares in January
2025 at a share price of $14.56 per share. A further $60,000,000 in Milestone Rights, or performance rights, is payable in
cash and/or in ordinary shares, upon achievement of certain clinical milestones. The purchase price also includes a
deferred amount payable of $3,472,000 (up to a maximum of $4,000,000 in equity) at the conclusion of a 15-month non-
fundamental indemnity period, which is subject to set-off in the instance we have substantiated claims against ImaginAb.
Upon achievement of specific key development and commercial milestones, Telix will pay up to a total of $185 million, a
portion of which may be paid in cash or equity at Telix’s election. Royalties are also payable on net sales in the low single
digits on a limited number of platform and early-stage products after the first four products have been developed, as
well as single-digit sublicense fees, as applicable.
Upfront equity consideration was subject to voluntary escrow (lock-up/leak-out) restrictions with equal tranches being
released from escrow 60, 90 and 120 days after closing. The ImaginAb Transaction agreements contain comprehensive
fundamental and non-fundamental representations, indemnity protections, and specific non-compete and non-
solicitation restrictions for ImaginAb, among other standard terms for such agreements.
127
Government Regulation and Product Approval
Government authorities in the U.S., at the federal, state and local level, and in other countries and jurisdictions, including
the European Union ("EU") extensively regulate, among other things, the research, development, testing, manufacture,
quality control, approval, packaging, storage, recordkeeping, labeling, advertising, promotion, distribution, marketing,
sales, pricing, reimbursement, post-approval monitoring and reporting, and import and export of pharmaceutical
products. The processes for obtaining regulatory approvals in the U.S. and in foreign countries and jurisdictions, along
with subsequent compliance with applicable statutes and regulations and other regulatory authorities, require the
expenditure of substantial time and financial resources. The regulatory requirements applicable to product development,
approval and marketing are subject to change, and regulations and administrative guidance often are revised or
reinterpreted by government agencies in ways that may have a significant impact on our business.
Review and Approval of Drugs and Biologics in the U.S.
In the U.S., the FDA approves and regulates drugs under the FDCA and related regulations. Biological products are
licensed for marketing under the Public Health Service Act ("PHSA") and subject to regulation under the FDCA and
related regulations. Pursuant to Section 3621 of the Consolidated Appropriations Act of 2023, which was signed into law
on December 29, 2022, contrast agents and radioactive pharmaceuticals are regulated as drugs or biologics.
A company, institution, or organization which takes responsibility for the initiation and management of a clinical
development program for such products, and for their regulatory approval, is typically referred to as a sponsor. A
sponsor seeking approval to market and distribute a new drug or biological product in the U.S. must typically secure the
following:
•Completion of preclinical laboratory tests in compliance with the FDA’s good laboratory practice ("GLP") standards
and applicable regulations;
•Design of a clinical protocol and submission to the FDA of an Investigational New Drug Application ("IND"), which
must take effect before human clinical trials may begin;
•Approval by an IRB representing each clinical site before each clinical trial may be initiated;
•Performance of adequate and well-controlled human clinical trials in accordance with GCPs to establish the safety
and efficacy of the proposed drug product for each proposed indication or with respect to biologics, the safety,
purity and potency of the product candidate for each proposed indication;
•Submission to the FDA of an NDA, for a drug candidate product and a BLA for a biological product requesting
marketing for one or more proposed indications;
•Review of the request for approval by an FDA advisory committee, where appropriate or if applicable;
•Completion of one or more FDA inspections of the manufacturing facility or facilities at which the product, or
components thereof, are produced to assess compliance with cGMPs to assure the product’s identity, strength,
quality and purity;
•Completion of FDA audits of clinical trial sites to assure compliance with GCPs and the integrity of the clinical data;
•Payment of user fees pursuant to the PDUFA;
•Securing FDA approval of the NDA or BLA; and
•Compliance with any post-approval requirements, including the potential requirement to implement a REMS and the
potential requirement to conduct post-approval studies.
Preclinical Studies
Before a sponsor begins testing a drug or biologic compound with potential diagnostic or therapeutic value in humans,
the product candidate enters the preclinical testing stage. Preclinical studies include laboratory evaluation of the purity
and stability of the manufactured substance or active pharmaceutical ingredient and the formulated product, as well as in
vitro and animal studies to assess the safety and activity of the product candidate for initial testing in humans and to
establish a rationale for therapeutic use. These studies are generally referred to as IND-enabling studies. The conduct of
preclinical studies is subject to federal regulations and requirements, including GLP standards and regulations and the
U.S. Department of Agriculture’s Animal Welfare Act, if applicable. With passage of the FDA’s Modernization Act 2.0 in
December 2022, Congress eliminated provisions in both the FDCA and the PHSA that required animal testing in support
of an NDA or BLA. While animal testing may still be conducted, the FDA was authorized to rely on alternative non-clinical
tests, including cell-based assays, micro-physiological systems, or bio-printed or computer models. Some long-term
preclinical testing, such as animal tests of reproductive adverse events and carcinogenicity, and long-term toxicity
studies, may continue after the IND is submitted.
128
The IND and IRB Processes
An IND is a request for FDA authorization to administer a product candidate to humans. Such authorization must be
secured prior to interstate shipment and administration of any new drug or biologic that is not the subject of an approved
NDA or BLA. In support of a request for an IND, sponsors must submit a protocol for each clinical trial and any
subsequent protocol amendments must be submitted to the FDA as part of the IND. In addition, the results of the
preclinical tests, together with manufacturing information, analytical data, any available clinical data or literature and
plans for clinical trials, among other things, are submitted to the FDA as part of an IND. The FDA requires a 30-day
waiting period after the filing of each IND before clinical trials may begin. This waiting period is designed to allow the FDA
to review the IND to determine whether human research subjects and patients will be exposed to unreasonable health
risks. At any time during this 30-day period, or thereafter, the FDA may raise concerns or questions about the conduct of
the trials as outlined in the IND and impose a clinical hold or partial clinical hold. In this case, the IND sponsor and the
FDA must resolve any outstanding concerns before clinical trials can begin or continue, if the clinical hold is initiated after
the study has begun. The FDA’s primary objectives in reviewing an IND are to assure the safety and rights of patients and
to help assure that the quality of the investigation will be adequate to permit an evaluation of the drug’s effectiveness
and safety and of the biological product’s safety, purity and potency.
A clinical hold is an order issued by the FDA to the sponsor to delay a proposed clinical investigation or to suspend an
ongoing investigation. A partial clinical hold is a delay or suspension of only part of the clinical protocol or protocols
under the IND. For example, a specific protocol or part of a protocol is not allowed to proceed, while other protocols or
parts of the protocols may do so. Following issuance of a clinical hold or partial clinical hold, an investigation may only
resume after the FDA has notified the sponsor that the investigation may proceed. The FDA will base that determination
on information provided by the sponsor correcting the deficiencies previously cited or otherwise demonstrating to the
satisfaction of the FDA that the investigation can proceed.
A sponsor may choose, but is not required, to conduct a foreign clinical study under an IND. When a foreign clinical study
is conducted under an IND, all IND requirements must be met unless waived. When a foreign clinical study is not
conducted under an IND, the sponsor must ensure that the study complies with certain regulatory requirements of the
FDA in order to use the study as support for an IND or application for marketing approval in the U.S.
The FDA’s regulations are intended to help ensure the protection of human subjects enrolled in non-IND foreign clinical
studies, as well as the quality and integrity of the resulting data. They further help ensure non-IND foreign studies are
conducted in a manner comparable to that required for IND studies.
In addition to the foregoing IND requirements, an IRB representing each institution participating in the clinical trial must
review and approve the plan for any clinical trial before it commences at that institution, and the IRB must conduct
continuing review and reapprove the trial at least annually. The IRB must review and approve, among other things, the
trial protocol and informed consent information to be provided to trial subjects. An IRB must operate in compliance with
FDA regulations. An IRB can suspend or terminate approval of a clinical trial at its institution, or an institution it
represents, if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the product
candidate has been associated with unexpected serious harm to patients.
Additionally, some trials are overseen by an independent group of qualified experts organized by the trial sponsor, known
as a data monitoring committee. This group provides authorization for whether a trial may move forward at designated
check points based on access only the group maintains to available data from the trial. Suspension or termination of
development during any phase of clinical trials can occur if it is determined the participants or patients are being
exposed to an unacceptable health risk.
Human Clinical Studies in Support of an NDA or BLA
Clinical trials involve the administration of the investigational product to human subjects under the supervision of
qualified investigators in accordance with GCP requirements, which include, among other things, the requirement that all
research subjects provide their informed consent in writing before their participation in any clinical trial. Clinical trials are
conducted under written trial protocols detailing, among other things, the inclusion and exclusion criteria, the objectives
of the trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated.
The clinical investigation of an investigational drug or biological product is generally divided into four phases. Although
the phases are usually conducted sequentially, they may overlap or be combined. The four phases of an investigation are
as follows:
•Phase 1: Studies include the initial introduction of an investigational new drug or biological product into humans.
These studies are designed to evaluate the safety, dosage tolerance, metabolism and pharmacologic actions of the
investigational drug or biological product in humans, any adverse events associated with increasing doses, and if
possible, to gain early evidence on effectiveness.
•Phase 2: Includes controlled clinical trials conducted to preliminarily or further evaluate the effectiveness of the
investigational drug or biological product for a particular indication(s) in patients with the disease or condition under
trial, to determine dosage tolerance and optimal dosage, and to identify possible adverse events and safety risks
associated with the drug or biological product. Phase 2 clinical trials are typically well-controlled, closely monitored,
and conducted in a limited patient population.
129
•Phase 3: Clinical trials are generally controlled clinical trials conducted in an expanded patient population generally
at geographically dispersed clinical trial sites. They are performed after preliminary evidence suggesting
effectiveness of the drug or biological product has been obtained, and are intended to further evaluate dosage,
clinical effectiveness and safety, to establish the overall benefit-risk relationship of the investigational drug or
biological product, and to provide an adequate basis for product approval.
•Phase 4: Post-approval studies may be conducted after initial marketing approval. These studies are used to gain
additional experience from the treatment of patients in the intended therapeutic indication.
A clinical trial may combine the elements of more than one phase and the FDA often requires more than one Phase 3 trial
to support marketing approval of a product candidate. A company’s designation of a clinical trial as being of a particular
phase is not necessarily indicative the study will be sufficient to satisfy the FDA requirements of that phase because this
determination cannot be made until the protocol and data have been submitted to and reviewed by the FDA. Generally,
pivotal trials are Phase 3 trials, but they may be Phase 2 trials if the design provides a well-controlled and reliable
assessment of clinical benefit, particularly in an area of unmet medical need.
In March 2022, the FDA released final guidance entitled “Expansion Cohorts: Use in First-In-Human Clinical Trials to
Expedite Development of Oncology Drugs and Biologics,” which outlines how developers can utilize an adaptive trial
design commonly referred to as a seamless trial design in early stages of oncology biological product development (i.e.,
the first-in-human clinical trial) to compress the traditional three phases of trials into one continuous trial called an
expansion cohort trial. Information to support the design of individual expansion cohorts are included in IND applications
and assessed by FDA. Expansion cohort trials can potentially bring efficiency to biological product development and
reduce developmental costs and time.
In December 2022, with the passage of FDORA, Congress required sponsors to develop and submit a DAP for each
Phase 3 clinical trial or any other “pivotal study” of a new drug or biological product. These plans are meant to encourage
the enrollment of more diverse patient populations in late-stage clinical trials of FDA-regulated products. Specifically,
action plans must include the sponsor’s goals for enrollment, the underlying rationale for those goals, and an explanation
of how the sponsor intends to meet them. In June 2024, as mandated by FDORA, the FDA issued draft guidance outlining
the general requirements for DAPs. Unlike most guidance documents issued by the FDA, the DAP guidance when
finalized will have the force of law because FDORA specifically dictates the form and manner for submission of DAPs are
specified in FDA guidance. On January 27, 2025, in response to an Executive Order issued by President Trump on
January 21, 2025, on Diversity, Equity and Inclusion programs, the FDA removed this draft guidance from its website.
This action raises questions about the applicability of statutory obligations to submit DAPs and the agency’s current
thinking on best practices for clinical development.
In June 2023, the FDA issued draft guidance with updated recommendations for GCPs aimed at modernizing the design
and conduct of clinical trials. The updates are intended to help pave the way for more efficient clinical trials to facilitate
the development of medical products. The draft guidance is adopted from the International Council for Harmonisation of
Technical Requirements for Pharmaceuticals for Human Use’s ("ICH's"), recently updated E6(R3) draft guideline
developed to enable the incorporation of rapidly developing technological and methodological innovations into the
clinical trial enterprise. In addition, the FDA issued draft guidance outlining recommendations for the implementation of
decentralized clinical trials.
Finally, sponsors of clinical trials are required to register and disclose certain clinical trial information on a public registry
(clinicaltrials.gov) maintained by the U.S. National Institutes of Health ("NIH"). In particular, information related to the
product, patient population, phase of investigation, study sites and investigators and other aspects of the clinical trial is
made public as part of the registration of the clinical trial. The failure to submit clinical trial information to
clinicaltrials.gov, as required, is a prohibited act under the FDCA with violations subject to potential civil monetary
penalties of up to US$10,000 for each day the violation continues. To date, the FDA has issued six notices of non-
compliance, thereby signaling the government’s willingness to begin enforcing these requirements against non-compliant
clinical trial sponsors. These notices of non-compliance did not result in civil monetary penalties.
Concurrent with clinical trials, companies often complete additional animal studies and must also develop additional
information about the chemistry and physical characteristics of the candidate product as well as finalize a process for
manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process
must be capable of consistently producing quality batches of the product candidate and, among other things, must
develop methods for testing the identity, strength, quality, purity, and potency of the final product candidate.
Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate
the product candidate does not undergo unacceptable deterioration over its shelf life.
Clinical Studies Outside the U.S. in Support of FDA Approval
In connection with our clinical development programs, we are conducting trials at sites outside the U.S. When a foreign
clinical trial is conducted under an IND, all IND requirements must be met unless waived. When a foreign clinical trial is
not conducted under an IND, the sponsor must ensure the trial complies with certain regulatory requirements of the FDA
in order to use the trial as support for an IND or application for marketing approval. Specifically, the trials must be
conducted in accordance with GCP, including undergoing review and receiving approval by an independent ethics
committee ("IEC") and seeking and receiving informed consent from subjects. GCP requirements encompass both ethical
and data integrity standards for clinical trials. The FDA’s regulations are intended to help ensure the protection of human
130
subjects enrolled in non-IND foreign clinical trials, as well as the quality and integrity of the resulting data. They further
help ensure that non-IND foreign trials are conducted in a manner comparable to that required for IND trials.
The acceptance by the FDA of trial data from clinical trials conducted outside the U.S. in support of U.S. approval may be
subject to certain conditions or may not be accepted at all. In cases where data from foreign clinical trials are intended to
serve as the sole basis for marketing approval in the U.S., the FDA will generally not approve the application on the basis
of foreign data alone unless (i) the data are applicable to the U.S. population and U.S. medical practice; (ii) the trials were
performed by clinical investigators of recognized competence and pursuant to GCP regulations; and (iii) the data may be
considered valid without the need for an on-site inspection by the FDA, or if the FDA considers such inspection to be
necessary, the FDA is able to validate the data through an on-site inspection or other appropriate means.
In addition, even where the foreign trial data are not intended to serve as the sole basis for approval, the FDA will not
accept the data as support for an application for marketing approval unless the trial is well-designed and well-conducted
in accordance with GCP requirements and the FDA is able to validate the data from the trial through an onsite inspection
if deemed necessary. Many foreign regulatory authorities have similar approval requirements. In addition, such foreign
trials are subject to the applicable local laws of the foreign jurisdictions where the trials are conducted.
Interactions with the FDA During the Clinical Development Program
Following the clearance of an IND and the commencement of clinical trials, the sponsor will continue to have interactions
with the FDA. Progress reports detailing the results of clinical trials must be submitted annually within 60 days of the
anniversary dates that the IND went into effect and more frequently if serious adverse events occur.
These reports must include a development safety update report ("DSUR"). In addition, IND safety reports must be
submitted to the FDA for any of the following: serious and unexpected suspected adverse reactions; findings from other
studies or animal or in vitro testing that suggest a significant risk in humans exposed to the product; and any clinically
important increase in the occurrence of a serious suspected adverse reaction over that listed in the protocol or
investigator brochure. Phase 1, Phase 2, and Phase 3 clinical trials may not be completed successfully within any
specified period, or at all. The FDA will typically inspect one or more clinical sites to assure compliance with GCP and the
integrity of the clinical data submitted. With passage of FDORA, Congress clarified FDA’s authority to conduct
inspections by expressly permitting inspection of facilities involved in the preparation, conduct, or analysis of clinical and
non-clinical studies submitted to FDA as well as other persons holding study records or involved in the study process.
In addition, sponsors are given opportunities to meet with the FDA at certain points in the clinical development program.
Specifically, sponsors may meet with the FDA prior to the submission of an IND ("Pre-IND meeting"), at the end of Phase
2 clinical trial ("EOP2 meeting") and before an NDA is submitted ("Pre-NDA meeting"). Meetings at other times may also
be requested. There are five types of meetings that occur between sponsors and the FDA.
Type A meetings are those that are necessary for an otherwise stalled product development program to proceed or to
address an important safety issue. Type B meetings include pre-IND and pre-NDA meetings as well as end of phase
meetings such as EOP2 meetings. A Type C meeting is any meeting other than a Type A or Type B meeting regarding the
development and review of a product, including for example meetings to facilitate early consultations on the use of a
biomarker as a new surrogate endpoint that has never been previously used as the primary basis for product approval in
the proposed context of use. A type D meeting is focused on a narrow set of issues (should be limited to no more than 2
focused topics) and should not require input from more than 3 disciplines or Divisions. Finally, INTERACT meetings are
intended for novel products and development programs that present unique challenges in the early development of an
investigational product.
The FDA has indicated that its responses, as conveyed in meeting minutes and advice letters, only constitute mere
recommendations and/or advice made to a sponsor and, as such, sponsors are not bound by such recommendations
and/or advice. Nonetheless, from a practical perspective, a sponsor’s failure to follow the FDA’s recommendations for
design of a clinical program may put the program at significant risk of failure.
Manufacturing and Other Regulatory Requirements
Concurrently with clinical trials, sponsors usually complete additional animal safety studies, develop additional
information about the chemistry and physical characteristics of the product candidate, and finalize a process for
manufacturing commercial quantities of the product candidate in accordance with cGMP requirements. The
manufacturing process must be capable of consistently producing quality batches of the product candidate and, among
other criteria, the sponsor must develop methods for testing the identity, strength, quality, and purity of the finished
product. Additionally, appropriate packaging must be selected and tested, and stability studies must be conducted to
demonstrate that the product candidate does not undergo unacceptable deterioration over its shelf life.
Specifically, the FDA’s regulations require that pharmaceutical products be manufactured in specific approved facilities
and in accordance with cGMPs. The cGMP regulations include requirements relating to organization of personnel,
buildings and facilities, equipment, control of components and product containers and closures, production and process
controls, packaging and labeling controls, holding and distribution, laboratory controls, records and reports, and returned
or salvaged products. Manufacturers and other entities involved in the manufacture and distribution of approved
pharmaceuticals are required to register their establishments with the FDA and some state agencies, and they are
subject to periodic unannounced inspections by the FDA for compliance with cGMPs and other requirements. The
131
PREVENT Pandemics Act, which was enacted in December 2022, clarifies that foreign drug manufacturing
establishments are subject to registration and listing requirements even if a drug undergoes further manufacture,
preparation, propagation, compounding, or processing at a separate establishment outside the U.S. prior to being
imported or offered for import into the U.S.
Pediatric Studies
Under PREA, an application or supplement thereto must contain data that are adequate to assess the safety and
effectiveness of the product for the claimed indications in all relevant pediatric subpopulations, and to support dosing
and administration for each pediatric subpopulation for which the product is safe and effective. Sponsors must also
submit an initial Pediatric Study Plan ("PSP") prior to the assessment data. The PSP must contain an outline of the
proposed pediatric study or studies the sponsor plans to conduct, including study objectives and design, any deferral or
waiver requests and other information required by regulation. The sponsor, the FDA, and the FDA’s internal review
committee must then review the information submitted, consult with each other and agree upon a final plan. The FDA or
the sponsor may request an amendment to the plan at any time.
The FDA may, on its own initiative or at the request of the sponsor, grant deferrals for submission of some or all pediatric
data until after approval of the product for use in adults, or full or partial waivers from the pediatric data requirements.
A deferral may be granted for several reasons, including a finding that the product or therapeutic candidate is ready for
approval for use in adults before pediatric trials are complete or that additional safety or effectiveness data needs to be
collected before the pediatric trials begin. The FDA is required to send a PREA Non-Compliance letter to sponsors who
have failed to submit their pediatric assessments required under PREA, have failed to seek or obtain a deferral or deferral
extension or have failed to request approval for a required pediatric formulation. Unless otherwise required by regulation,
the pediatric data requirements do not apply to products with orphan designation, although the FDA has recently taken
steps to limit what it considers abuse of this statutory exemption.
The Food and Drug Administration Reauthorization Act of 2017 ("FDARA") also established new requirements to govern
certain molecularly targeted cancer indications. Any company that submits an application three years after the date of
enactment of that statute must submit pediatric assessments with the application if the product is intended for the
treatment of an adult cancer and is directed at a molecular target that the FDA determines to be substantially relevant to
the growth or progression of a pediatric cancer. The investigation must be designed to yield clinically meaningful
pediatric study data regarding the dosing, safety and preliminary efficacy to inform pediatric labeling for the product.
Section 505(b)(2) NDAs
NDAs for most new drug products are based on two full clinical studies which must contain substantial evidence of the
safety and efficacy of the proposed new product for the proposed use. These applications are submitted under Section
505(b)(1) of the FDCA. The FDA is, however, authorized to approve an alternative type of NDA under Section 505(b)(2)
of the FDCA. This type of application allows the sponsor to rely, in part, on the FDA’s previous findings of safety and
efficacy for a similar product, or published literature. Specifically, Section 505(b)(2) applies to NDAs for a drug for which
the investigations made to show whether or not the drug is safe for use and effective in use and relied upon by the
sponsor for approval of the application “were not conducted by or for the applicant and for which the applicant has not
obtained a right of reference or use from the person by or for whom the investigations were conducted.”
Section 505(b)(2) thus authorizes the FDA to approve an NDA based on safety and effectiveness data that were not
developed by the applicant. NDAs filed under Section 505(b)(2) may provide an alternate and potentially more
expeditious pathway to FDA approval for new or improved formulations or new uses of previously approved products.
If the 505(b)(2) applicant can establish that reliance on the FDA’s previous approval is scientifically appropriate, the
applicant may eliminate the need to conduct certain preclinical or clinical studies of the new product. The FDA may also
require companies to perform additional studies or measurements to support the change from the approved product. The
FDA may then approve the new drug candidate for all or some of the label indications for which the referenced product
has been approved as well as for any new indication sought by the Section 505(b)(2) applicant.
Fast Track, Breakthrough Therapy and Priority Review Designations
The FDA is authorized to designate certain products for expedited review if they are intended to address an unmet
medical need in the treatment of a serious or life-threatening disease or condition. These programs include fast track
designation, breakthrough therapy designation and priority review designation. None of these expedited programs
changes the standards for approval but each may help expedite the development or approval process governing product
candidates.
Specifically, the FDA may designate a product for fast track review if it is intended, whether alone or in combination with
one or more other products, for the treatment of a serious or life-threatening disease or condition, and it demonstrates
the potential to address unmet medical needs for such a disease or condition. For fast track products, sponsors may
have greater interactions with the FDA and the FDA may initiate review of sections of a fast track product’s application
before the application is complete. This rolling review may be available if the FDA determines, after preliminary
evaluation of clinical data submitted by the sponsor, that a fast track product may be effective. The sponsor must also
provide, and the FDA must approve, a schedule for the submission of the remaining information and the sponsor must
132
pay applicable user fees. However, the FDA’s time period goal for reviewing a fast track application does not begin until
the last section of the application is submitted. In addition, the fast track designation may be withdrawn by the FDA if the
FDA believes that the designation is no longer supported by data emerging in the clinical trial process.
Second, a product may be designated as a breakthrough therapy if it is intended, either alone or in combination with one
or more other products, to treat a serious or life-threatening disease or condition and preliminary clinical evidence
indicates that the product may demonstrate substantial improvement over existing therapies on one or more clinically
significant endpoints, such as substantial treatment effects observed early in clinical development. The FDA may take
certain actions with respect to breakthrough therapies, including holding meetings with the sponsor throughout the
development process; providing timely advice to the product sponsor regarding development and approval; involving
more senior staff in the review process; assigning a cross-disciplinary project lead for the review team; and taking other
steps to help the sponsor design the clinical trials in an efficient manner.
Third, the FDA may designate a product for priority review if it is a product that treats a serious condition and, if
approved, would provide a significant improvement in safety or effectiveness. The FDA determines, on a case-by-case
basis, whether the proposed product represents a significant improvement when compared with other available
therapies. Significant improvement may be illustrated by evidence of increased effectiveness in the treatment of a
condition, elimination or substantial reduction of a treatment-limiting product reaction, documented enhancement of
patient compliance that may lead to improvement in serious outcomes, and evidence of safety and effectiveness in a
new subpopulation. A priority designation is intended to direct overall attention and resources to the evaluation of such
applications, and to shorten the FDA’s goal for taking action on a marketing application from ten months to six months.
Accelerated Approval Pathway
The FDA may grant accelerated approval to a product for a serious or life-threatening condition that provides meaningful
therapeutic advantage to patients over existing treatments based upon a determination that the product has an effect on
a surrogate endpoint that is reasonably likely to predict clinical benefit. The FDA may also grant accelerated approval for
such a condition when the product has an effect on an intermediate clinical endpoint that can be measured earlier than
an effect on irreversible morbidity or mortality ("IMM") and that is reasonably likely to predict an effect on irreversible
morbidity or mortality or other clinical benefit, taking into account the severity, rarity or prevalence of the condition and
the availability or lack of alternative treatments. Products granted accelerated approval must meet the same statutory
standards for safety and effectiveness as those granted traditional approval.
For the purposes of accelerated approval, a surrogate endpoint is a marker, such as a laboratory measurement,
radiographic image, physical sign or other measure that is thought to predict clinical benefit, but is not itself a measure of
clinical benefit. Surrogate endpoints can often be measured more easily or more rapidly than clinical endpoints. An
intermediate clinical endpoint is a measurement of a therapeutic effect that is considered reasonably likely to predict the
clinical benefit of a drug, such as an effect on IMM.
The accelerated approval pathway is usually contingent on a sponsor’s agreement to conduct, in a diligent manner,
additional post-approval confirmatory studies to verify and describe the product’s clinical benefit. As a result, a product
candidate approved on this basis is subject to rigorous post-marketing compliance requirements, including the
completion of Phase 4 or post-approval clinical trials to confirm the effect on the clinical endpoint. Failure to conduct
required post-approval studies, or confirm a clinical benefit during post-marketing studies, would allow the FDA to
initiate expedited proceedings to withdraw approval of the product. All promotional materials for product candidates
approved under accelerated regulations are subject to prior review by the FDA.
With passage of FDORA in December 2022, Congress modified certain provisions governing accelerated approval of drug
and biologic products. Specifically, the new legislation authorized the FDA to require a sponsor to have its confirmatory
clinical trial underway before accelerated approval is awarded and to submit progress reports on its post-approval
studies to FDA every six months until the study is completed. Moreover, FDORA established expedited procedures
authorizing FDA to withdraw an accelerated approval if certain conditions are met, including where a required
confirmatory study fails to verify and describe the predicted clinical benefit or where evidence demonstrates the product
is not shown to be safe or effective under the conditions of use. The FDA may also use such procedures to withdraw an
accelerated approval if a sponsor fails to conduct any required post-approval study of the product with due diligence,
including with respect to “conditions specified by the Secretary.” The new procedures include the provision of due notice
and an explanation for a proposed withdrawal, and opportunities for a meeting with the Commissioner or the
Commissioner’s designee and a written appeal, among other things.
More recently, in March 2023, the FDA issued draft guidance that outlines its current thinking and approach to
accelerated approval. The agency indicated that the accelerated approval pathway is commonly used for approval of
oncology drugs due to the serious and life-threatening nature of cancer. Although single-arm trials have been commonly
used to support accelerated approval, a randomized controlled trial is the preferred approach as it provides a more
robust efficacy and safety assessment and allows for direct comparisons to an available therapy. To that end, the FDA
outlined considerations for designing, conducting, and analyzing data for trials intended to support accelerated
approvals of oncology therapeutics. Subsequently, in December 2024 and January 2025, the FDA issued additional draft
guidances relating to accelerated approval. These guidances describe FDA’s latest thinking on what it means to conduct
a confirmatory trial with due diligence and how the agency plans to interpret whether such a study needs to be
underway at the time of approval. While these guidances are currently only in draft form and will ultimately not be legally
133
binding even when finalized, sponsors typically observe the FDA’s guidance closely to ensure that their investigational
products qualify for accelerated approval.
FDA Commissioner’s National Priority Review Voucher (CNPV) Pilot Program
In June 2025, the FDA announced the CNPV pilot program which was designed to accelerate the development and
review of certain drugs and biologics that are aligned with U.S. national health priorities, such as addressing a U.S. public
health crisis, developing more innovative cures for the American people, addressing a large unmet medical need,
onshoring drug development and manufacturing to advance the health interests of Americans and strengthen U.S. supply
chain resiliency, and increasing affordability. The FDA has stated that voucher recipients will receive a decision with
respect to an application on an accelerated basis, as well as enhanced communication with review staff throughout the
review process. The FDA expects the CNPV program to accelerate the application review timeline from 10-12 months to
1-2 months by convening a multidisciplinary team of physicians and scientists for a team-based review, interacting
frequently with the sponsor of the application to clarify questions, and completing review of the application concurrently.
The FDA retains full discretion to extend the review window if the data or application components submitted are
insufficient or incomplete, if the results of the pivotal trial(s) are ambiguous, or if the review is particularly complex. The
FDA has indicated the CNPV program does not change the FDA’s rigorous safety and efficacy standards for review and
approval. In late 2025, FDA began issuing approvals for drugs with CNPVs within the 1-2 month review timeframe. The
CNPV program is new, limited in scope, and subject to evolving guidance, and available FDA resources. The FDA retains
broad discretion to modify the criteria, processes, or benefits of the program and may rescind participation or alter
timelines or the intended benefits at any time. Adding to the uncertainty, concerns have been raised regarding the
legality of the CNPV program.
Submission and Review of an NDA or BLA by the FDA
Assuming successful completion of the required clinical testing, the results of the preclinical studies and clinical trials,
along with information relating to the product’s chemistry, manufacturing, controls, safety updates, patent information,
abuse information, and proposed labeling, are submitted to the FDA as part of an application requesting approval to
market the product candidate for one or more indications. Data may come from company-sponsored clinical trials
intended to test the safety and efficacy of a product’s use or from a number of alternative sources, including studies
initiated by investigators. To support marketing approval, the data submitted must be sufficient in quality and quantity to
establish the safety and efficacy of a drug product, and potency, purity and safety of a biologic product, to the
satisfaction of the FDA.
The fee required for the submission and review of an application under PDUFA is substantial (for example, for fiscal year
2026, this application fee is US$4,682,003), and the sponsor of an approved application is also subject to an annual
program fee, which for fiscal year 2025 is US$442,213 per eligible prescription product. These fees are typically adjusted
annually, and exemptions and waivers may be available under certain circumstances, such as where a waiver is
necessary to protect the public health, where the fee would present a significant barrier to innovation, or where the
sponsor is a small business submitting its first human drug application for review.
The FDA conducts a preliminary review of all applications within 60 days of receipt and must inform the sponsor at that
time or before whether an application is sufficiently complete to permit substantive review. In pertinent part, the FDA’s
regulations state that an application “shall not be considered as filed until all pertinent information and data have been
received” by the FDA. In the event that the FDA determines that an application does not satisfy this standard, it will issue
an RTF determination to the applicant. Typically, an RTF will be based on administrative incompleteness, such as clear
omission of information or sections of required information; scientific incompleteness, such as omission of critical data,
information, or analyses needed to evaluate safety and efficacy or provide adequate directions for use; or inadequate
content, presentation, or organization of information such that substantive and meaningful review is precluded. The FDA
may request additional information rather than accept an application for filing. In this event, the application must be
resubmitted with the additional information. The resubmitted application is also subject to review before the FDA accepts
it for filing.
After the submission is accepted for filing, the FDA begins an in-depth substantive review of the application. The FDA
reviews the application to determine, among other things, whether the proposed product is safe and effective for its
intended use, whether it has an acceptable purity profile, and whether the product is being manufactured in accordance
with cGMP. Under the goals and policies agreed to by the FDA under PDUFA, the FDA has ten months from the filing date
in which to complete its initial review of a standard application that is a new molecular entity, and six months from the
filing date for an application with “priority review.” The review process may be extended by the FDA for three additional
months to consider new information or in the case of a clarification provided by the sponsor to address an outstanding
deficiency identified by the FDA following the original submission. Despite these review goals, it is not uncommon for
FDA review of an application to extend beyond the PDUFA goal date.
In connection with its review of an application, the FDA may submit information requests to the sponsor and set
deadlines for responses thereto. The FDA will also conduct a pre-approval inspection of the manufacturing facilities for
the new product to determine whether the manufacturing processes and facilities comply with cGMPs. The FDA will not
approve the product unless it determines that the manufacturing processes and facilities are in compliance with cGMP
requirements and are adequate to assure consistent production of the product within required specifications. The FDA
also may inspect the sponsor and one or more clinical trial sites to assure compliance with IND and GCP requirements
and the integrity of the clinical data submitted to the FDA.
134
Moreover, the FDA will review a sponsor’s financial relationship with the principal investigators who conducted the
clinical trials in support of the NDA. That is because, under certain circumstances, principal investigators at a clinical trial
site may also serve as scientific advisors or consultants to a sponsor and receive compensation in connection with such
services. Depending on the level of that compensation and any other financial interest a principal investigator may have
in a sponsor, the sponsor may be required to report these relationships to the FDA. The FDA will then evaluate that
financial relationship and determine whether it creates a conflict of interest or otherwise affects the interpretation of the
trial or the integrity of the data generated at the principal investigator’s clinical trial site. If so, the FDA may exclude data
from the clinical trial site in connection with its determination of safety and efficacy of the investigational product.
Additionally, the FDA may refer an application, including applications for novel product candidates which present difficult
questions of safety or efficacy, to an advisory committee for review, evaluation, and recommendation as to whether the
application should be approved and under what conditions. Typically, an advisory committee is a panel of independent
experts, including clinicians and other scientific experts, that reviews, evaluates, and provides a recommendation as to
whether the application should be approved and under what conditions. The FDA is not bound by the recommendation of
an advisory committee, but it considers such recommendations when making final decisions on approval.
The FDA also may require submission of a REMS if it determines that a REMS is necessary to ensure that the benefits of
the product outweigh its risks and to assure the safe use of the product. The REMS could include medication guides,
physician communication plans, assessment plans, and/or elements to assure safe use, such as restricted distribution
methods, patient registries, or other risk minimization tools. The FDA determines the requirement for a REMS, as well as
the specific REMS provisions, on a case-by-case basis. If the FDA concludes a REMS is needed, the sponsor of the
application must submit a proposed REMS and the FDA will not approve the application without a REMS.
The FDA’s Decision on an NDA or BLA
After evaluating the application and all related information, including the advisory committee recommendations, if any,
and inspection reports of manufacturing facilities and clinical trial sites, the FDA may issue either a Complete Response
Letter ("CRL") or an approval letter. To reach this determination, the FDA must evaluate whether the expected benefits of
the proposed product outweigh its potential risks to patients. This “benefit-risk” assessment is informed by the body of
evidence about the product’s safety and efficacy in the NDA or BLA.
If the FDA decides not to license or approve the application, it will issue a CRL. A CRL will describe all of the deficiencies
that the FDA has identified in the application, except that where the FDA determines that the data supporting the
application are inadequate to support approval, the FDA may issue the CRL without first conducting required inspections,
testing submitted product lots (where applicable), and/or reviewing proposed labeling. In issuing the CRL, the FDA may
recommend actions that the applicant might take to place the application in condition for approval, including requests for
additional information or clarification. The FDA may delay or refuse approval of an application if applicable regulatory
criteria are not satisfied, require additional testing or information and/or require post-marketing testing and surveillance
to monitor safety or efficacy of a product. If a CRL is issued, the applicant will have one year to respond to the
deficiencies identified by the FDA, at which time the FDA can deem the application withdrawn or, in its discretion, grant
the applicant an additional six month extension to respond. For those seeking to challenge the FDA’s CRL decision, the
FDA has indicated that sponsors may request a formal hearing on the CRL or they may file a request for reconsideration
or a request for a formal dispute resolution. The FDA recently announced that, in the interest of transparency, it will
release CRLs to the public.
An approval letter, on the other hand, authorizes commercial marketing of the product with specific prescribing
information for specific indications. If the FDA approves a product, it may limit the approved indications for use for the
product, require that contraindications, warnings or precautions be included in the product labeling, require that post-
approval studies, including Phase 4 clinical trials, be conducted to further assess the drug’s safety after approval, require
testing and surveillance programs to monitor the product after commercialization, or impose other conditions, including
distribution restrictions or other risk management mechanisms, including REMS, which can materially affect the potential
market and profitability of the product. The FDA may prevent or limit further marketing of a product based on the results
of post-market studies or surveillance programs. After approval, many types of changes to the approved product, such
as adding new indications, manufacturing changes and additional labeling claims, are subject to further testing
requirements and FDA review and approval.
Under the Ensuring Innovation Act, which was signed into law in April 2021, the FDA must publish action packages
summarizing its decisions to approve new drug products within 30 days of approval of such products.
Post-Approval Regulation
Drugs and biologics manufactured or distributed pursuant to FDA approvals are subject to pervasive and continuing
regulation by the FDA, including, among other things, requirements relating to recordkeeping, periodic reporting, product
sampling and distribution, advertising and promotion and reporting of adverse experiences with the product. After
approval, most changes to the approved product, such as adding new indications or other labeling claims, are subject to
prior FDA review and approval. There also are continuing, annual user fee requirements for any marketed products and
the establishments at which such products are manufactured, as well as new application fees for supplemental
applications with clinical data.
135
In addition, manufacturers and other entities involved in the manufacture and distribution of approved products are
required to register their establishments with the FDA and state agencies, and are subject to periodic unannounced
inspections by the FDA and these state agencies for compliance with cGMP requirements. Changes to the manufacturing
process are strictly regulated and may require prior FDA approval before being implemented. FDA regulations also
require investigation and correction of any deviations from cGMPs and impose reporting and documentation
requirements upon the sponsor and any third-party manufacturers that the sponsor may decide to use. Accordingly,
manufacturers must continue to expend time, money, and effort in the area of production and quality control to maintain
cGMP compliance.
Once an approval is granted, the FDA may withdraw the approval if compliance with regulatory requirements and
standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously
unknown problems with a product, including adverse events of unanticipated severity or frequency, or with
manufacturing processes, or failure to comply with regulatory requirements, may result in revisions to the approved
labeling to add new safety information; imposition of post-market studies or clinical trials to assess new safety risks; or
imposition of distribution or other restrictions under a REMS program. Other potential consequences include, among
other things:
•restrictions on the marketing or manufacturing of the product, suspension of the approval, or complete withdrawal
of the product from the market or product recalls;
•fines, warning letters or untitled letters or holds on post-approval clinical trials;
•refusal of the FDA to approve pending NDAs, BLAs or supplements to approved NDAs or BLAs, or suspension or
revocation of product approvals;
•product seizure or detention, or refusal to permit the import or export of products; or
•injunctions or the imposition of civil or criminal penalties.
The FDA strictly regulates marketing, labeling, advertising and promotion of products that are placed on the market.
Products may be promoted only for the approved indications and in accordance with the provisions of the approved
label. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of a product in a
manner inconsistent with the approved labeling, and a company that is found to have improperly promoted a product
uses may be subject to significant liability.
It may be permissible, under very specific, narrow conditions, for a manufacturer to engage in nonpromotional, non-
misleading communication regarding off-label information, such as distributing scientific or medical journal information.
Moreover, with passage of the Pre-Approval Information Exchange Act ("PIE Act"), in December 2022, sponsors of
products that have not been approved may proactively communicate to payors certain information about products in
development to help expedite patient access upon product approval. Previously, such communications were permitted
under FDA guidance, but the new legislation explicitly provides protection to sponsors who convey certain information
about products in development to payors, including unapproved uses of approved products. In January 2025, the FDA
published final guidance outlining the agency’s non-binding policies governing the distribution of scientific information on
unapproved uses to healthcare providers. This final guidance calls for such communications to be truthful, non-
misleading, factual, and unbiased and include all information necessary for healthcare providers to interpret the
strengths and weaknesses and validity and utility of the information about the unapproved use.
If a company is found to have promoted a product improperly, it may become subject to adverse public relations and
administrative and judicial enforcement by the FDA, the Department of Justice or the Office of the Inspector General of
HHS, as well as state authorities. This could subject a company to a range of penalties that could have a significant
commercial impact, including civil and criminal fines and agreements that materially restrict the manner in which a
company promotes or distributes drug products.
Generic Drugs and Regulatory Exclusivity
In 1984, with passage of the Drug Price Competition and Patent Term Restoration Act of 1984, commonly known as the
Hatch-Waxman Act, Congress established an abbreviated regulatory scheme authorizing the FDA to approve generic
drugs that are shown to contain the same active ingredients as, and to be bioequivalent to, drugs previously approved by
the FDA pursuant to NDAs and it also enacted Section 505(b)(2). To obtain approval of a generic drug, a sponsor must
submit an ANDA to the FDA. In support of such applications, a generic manufacturer may rely on the preclinical and
clinical testing conducted for a drug product previously approved under an NDA, known as the reference listed drug
("RLD").
Specifically, in order for an ANDA to be approved, the FDA must find that the generic version is identical to the RLD with
respect to the active ingredients, the route of administration, the dosage form, the strength of the drug, and the
conditions of use of the drug. At the same time, the FDA must also determine that the generic drug is “bioequivalent” to
the innovator drug.
Under the statute, a generic drug is bioequivalent to a RLD if “the rate and extent of absorption of the drug do not show a
significant difference from the rate and extent of absorption of the listed drug.” Upon approval of an ANDA, the FDA
136
indicates whether the generic product is “therapeutically equivalent” to the RLD in its publication “Approved Drug
Products with Therapeutic Equivalence Evaluations,” also referred to as the “Orange Book.” Physicians and pharmacists
consider a therapeutic equivalent generic drug to be fully substitutable for the RLD.
Under the Hatch-Waxman Act, the FDA may not approve an ANDA or 505(b)(2) application until any applicable period of
non-patent exclusivity for the RLD has expired. The FDCA provides a period of five years of regulatory exclusivity for a
new drug containing a new chemical entity ("NCE"). For the purposes of this provision, the FDA has consistently taken
the position that an NCE is a drug that contains no active moiety that has previously been approved by the FDA in any
other NDA. This interpretation was confirmed with enactment of the Ensuring Innovation Act in April 2021. An active
moiety is the molecule or ion responsible for the physiological or pharmacological action of the drug substance. In cases
where such NCE exclusivity has been granted, a generic or follow-on drug application may not be filed with the FDA until
the expiration of five years unless the submission is accompanied by a Paragraph IV certification, in which case the
sponsor may submit its application four years following the original product approval.
The FDCA also provides for a period of three years of regulatory exclusivity if the NDA includes reports of one or more
new clinical investigations, other than bioavailability or bioequivalence studies, that were conducted by or for the
sponsor and are essential to the approval of the application. This three-year exclusivity period often protects changes to
a previously approved drug product, such as new indications, dosage forms, route of administration or combination of
ingredients. Three-year exclusivity would be available for a drug product that contains a previously approved active
moiety, provided the statutory requirement for a new clinical investigation is satisfied. Unlike five-year NCE exclusivity,
an award of three-year exclusivity does not block the FDA from accepting ANDAs or 505(b)(2) NDAs seeking approval
for generic versions of the drug as of the date of approval of the original drug product; rather, this three-year exclusivity
covers only the conditions of use associated with the new clinical investigations and, as a general matter, does not
prohibit the FDA from approving follow-on applications for drugs containing the original active ingredient.
Five-year and three-year regulatory exclusivity also will not delay the submission or approval of a traditional NDA filed
under Section 505(b)(1) of the FDCA; however, a sponsor submitting a traditional NDA would be required to conduct or
obtain a right of reference to all of the preclinical studies and adequate and well-controlled clinical trials necessary to
demonstrate safety and effectiveness.
As part of the submission of an NDA or certain supplemental applications, NDA sponsors are required to list with the FDA
each patent with claims that cover the sponsor’s product or an approved method of using the product. Upon approval of
a new drug, each of the patents listed in the application for the drug is then published in the Orange Book. The FDA’s
regulations governing patient listings were largely codified into law with enactment of the Orange Book Modernization
Act in January 2021. When an ANDA applicant files its application with the FDA, the applicant is required to certify to the
FDA concerning any patents listed for the reference product in the Orange Book.
Specifically, the ANDA applicant must certify that: (i) the required patent information has not been filed; (ii) the listed
patent has expired; (iii) the listed patent has not expired but will expire on a particular date and approval is sought after
patent expiration; and/or (iv) the listed patent is invalid or will not be infringed by the new product. Moreover, to the
extent that the Section 505(b)(2) NDA applicant is relying on studies conducted for an already approved product, the
applicant also is required to certify to the FDA concerning any patents listed for the NDA-approved product in the
Orange Book to the same extent that an ANDA applicant would.
If the generic drug or follow-on drug applicant does not challenge the innovator’s listed patents, the FDA will not approve
the ANDA or 505(b)(2) application until all the listed patents claiming the referenced product have expired. A certification
that the new generic product will not infringe the already approved product’s listed patents or that such patents are
invalid or unenforceable is called a Paragraph IV certification. If the ANDA applicant has provided a Paragraph IV
certification to the FDA, the applicant must also send notice of the Paragraph IV certification to the NDA owner and
patent holders once the ANDA has been accepted for filing by the FDA. The NDA owner and patent holders may then
initiate a patent infringement lawsuit in response to the notice of the Paragraph IV certification. The filing of a patent
infringement lawsuit within 45 days after the receipt of a Paragraph IV certification automatically prevents the FDA from
approving the ANDA or 505(b)(2) NDA until the earliest of 30 months after the receipt of the Paragraph IV notice,
expiration of the patent and a decision in the infringement case that is favorable to the ANDA or 505(b)(2) NDA applicant.
Biosimilars and Regulatory Exclusivity
The ACA, which was signed into law on March 23, 2010, included a subtitle called the Biologics Price Competition and
Innovation Act of 2009 ("BPCIA"). The BPCIA established a regulatory scheme authorizing the FDA to approve biosimilars
and interchangeable biosimilars. To date, the FDA has approved a number of biosimilars and several interchangeable
biosimilar products.
Under the BPCIA, a manufacturer may submit an application for licensure of a biologic product that is “biosimilar to” or
“interchangeable with” a previously approved biological product or “reference product.” In order for the FDA to approve a
biosimilar product, it must find that there are no clinically meaningful differences between the reference product and
proposed biosimilar product in terms of safety, purity, and potency. For the FDA to approve a biosimilar product as
interchangeable with a reference product, the FDA must find that the biosimilar product can be expected to produce the
same clinical results as the reference product, and (for products administered multiple times) that the biologic and the
reference biologic may be switched after one has been previously administered without increasing safety risks or risks of
diminished efficacy relative to exclusive use of the reference biologic. In December 2022, Congress clarified through
137
FDORA that the FDA may approve multiple first interchangeable biosimilar biological products so long as the products are
all approved on the first day on which such a product is approved as interchangeable with the reference product.
Under the BPCIA, an application for a biosimilar product may not be submitted to the FDA until four years following the
date of approval of the reference product. The FDA may not approve a biosimilar product until 12 years from the date on
which the reference product was approved. Even if a product is considered to be a reference product eligible for
exclusivity, another company could market a competing version of that product if the FDA approves a full BLA for such
product containing the sponsor’s own preclinical data and data from adequate and well-controlled clinical trials to
demonstrate the safety, purity and potency of their product. The BPCIA also created certain exclusivity periods for
biosimilars approved as interchangeable products. There have been recent government proposals to reduce the 12-year
reference product exclusivity period, but none have been enacted to date. At the same time, since passage of the BPCIA,
many states have passed laws or amendments to laws, which address pharmacy practices involving biosimilar products.
Orphan Drug Designation and Exclusivity
Under the Orphan Drug Act, the FDA may designate a drug or biologic product as an “orphan drug” if it is intended to
treat a rare disease or condition, generally meaning that it affects fewer than 200,000 individuals in the U.S., or more in
cases in which there is no reasonable expectation that the cost of developing and making a drug product available in the
U.S. for treatment of the disease or condition will be recovered from sales of the product. A company must request
orphan drug designation before submitting an NDA or BLA for the candidate product. If the request is granted, the FDA
will disclose the identity of the product candidate and its potential use. Orphan drug designation does not shorten the
regulatory review and approval process, although it does convey certain advantages such as tax benefits and user fee
exemptions.
If a product with orphan designation receives the first FDA approval for the disease or condition for which it has such
designation or for a select indication or use within the rare disease or condition for which it was designated, the product
generally will receive orphan drug exclusivity. Orphan drug exclusivity means that the FDA may not approve another
sponsor’s marketing application for the same drug or biologic for the same disease or condition for seven years, except
in certain limited circumstances. Orphan exclusivity does not block the approval of a different product for the same rare
disease or condition, nor does it block the approval of the same product for different indications. If a drug or biologic
designated as an orphan drug ultimately receives marketing approval for an indication broader than what was designated
in its orphan drug application, it may not be entitled to exclusivity.
Orphan exclusivity will not bar approval of another product under certain circumstances, including if a company with
orphan drug exclusivity is not able to meet market demand and in cases where a subsequent product with the same drug
or biologic for the same indication is shown to be clinically superior to the approved product on the basis of greater
efficacy or safety, or providing a major contribution to patient care.
In September 2021, the Court of Appeals for the 11th Circuit held that, for the purpose of determining the scope of
market exclusivity, the term “same disease or condition” in the statute means the designated “rare disease or condition”
and could not be interpreted by the FDA to mean the “indication or use.” Thus, the court concluded, orphan drug
exclusivity applies to the entire designated disease or condition rather than the “indication or use.” Although there have
been legislative proposals to overrule this decision, they have not been enacted into law. On January 23, 2023, the FDA
announced that, in matters beyond the scope of that court order, the FDA will continue to apply its existing regulations
tying orphan-drug exclusivity to the uses or indications for which the orphan drug was approved.
Pediatric Exclusivity
Pediatric exclusivity is another type of non-patent marketing exclusivity in the U.S. and, if granted, provides for the
attachment of an additional six months of regulatory exclusivity. For drug products, the six-month exclusivity may be
attached to the term of any existing patent or regulatory exclusivity. For biologic products, the six-month period may
only be attached to any existing regulatory exclusivities but not to any patent terms. This six-month exclusivity may be
granted if an NDA or BLA sponsor submits pediatric data that fairly respond to a written request from the FDA for such
data. The data do not need to show the product to be effective in the pediatric population studied; rather, if the clinical
trial is deemed to fairly respond to the FDA’s request, the additional protection is granted. If reports of requested
pediatric studies are submitted to and accepted by the FDA within the statutory time limits, whatever statutory or
regulatory periods of non-patent exclusivity for drugs and biologics, or patent protection that covers a drug product, are
extended by six months. This is not a patent term extension, but it effectively extends the regulatory period during which
the FDA cannot approve another application.
Patent Term Restoration and Extension
A patent claiming a new product may be eligible for a limited patent term extension under the Hatch-Waxman Act, which
permits a patent restoration of up to five years for patent term lost during product development and the FDA regulatory
review. The restoration period granted on a patent covering a product is typically one-half the time between the
effective date of the IND approval and the submission date of an application, plus the time between the submission date
of the regulatory approval application and the ultimate approval date, depending on the amount of that time period in
which the subject patent was granted. Patent term restoration cannot be used to extend the remaining term of a patent
past a total of 14 years from the product’s approval date. Only one patent applicable to an approved product is eligible
for the extension, and the application for the extension must be submitted prior to the expiration of the patent in
138
question. A patent that covers multiple products for which approval is sought can only be extended in connection with
one of the approvals. The USPTO reviews and approves the application for any patent term extension or restoration in
consultation with the FDA.
U.S. Regulation of Medical Devices
The FDCA defines a medical device in pertinent part to include any instrument, apparatus, implement, machine,
contrivance, implant, in vitro reagent, or other similar or related article, including a component part, or accessory,
intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of
disease, in man or other animals, or to affect the structure or function of the body. Among other things, pursuant to the
FDCA and its implementing regulations, the FDA regulates the research, testing, manufacturing, safety, labeling, storage,
recordkeeping, premarket clearance or approval, marketing and promotion and sales and distribution of medical devices
in the U.S. In addition to traditional devices, like surgical tools, the FDA regulates certain software, including artificial
intelligence and machine learning algorithms, as medical devices depending on their intended use.
Device Classification
The FDA categorizes medical devices into one of three classes—Class I, II, or III—based on the risks presented by the
device and the regulatory controls necessary to provide a reasonable assurance of the device’s safety and effectiveness.
Class I includes devices with the lowest risk to the patient and are those for which safety and effectiveness can be
assured by adherence to the FDA’s General Controls for medical devices, which include compliance with the applicable
portions of the Quality System Regulation ("QSR"), facility registration and product listing, reporting of adverse medical
events or certain malfunctions, and truthful and non-misleading labeling, advertising, and promotional materials. Class II
devices are subject to the FDA’s General Controls, and special controls as deemed necessary by the FDA to ensure the
safety and effectiveness of the device. Special controls are established by the FDA for a specific device type and often
include specific labeling provisions, performance metrics, and other types of controls that mitigate risks of the device.
Devices deemed by the FDA to pose the greatest risks, such as life sustaining, life supporting or some implantable
devices, or devices that have a new intended use, or use advanced technology that is not substantially equivalent to that
of a legally marketed device, are placed in Class III, requiring approval of a PMA. Some pre-amendment devices are
unclassified, but are subject to the FDA’s premarket notification and clearance process in order to be commercially
distributed.
PMA Pathway
Class III devices generally require PMA approval before they can be marketed. Obtaining PMA approval requires the
submission of “valid scientific evidence” to the FDA to support a finding of a reasonable assurance of the safety and
effectiveness of the device. A PMA must provide complete analytical and clinical performance data and also information
about the device and its components regarding, among other things, device design, manufacturing and labeling.
Following receipt of a PMA, the FDA determines whether the application is sufficiently complete to permit a substantive
review. If the FDA accepts the application for review, it has 180 days under the FDCA to complete its review of a PMA,
although in practice, FDA’s review often takes significantly longer, and can take up to several years. An advisory panel of
experts from outside the FDA may be convened to review and evaluate the application and provide recommendations to
the FDA as to the approvability of the device. The FDA may or may not accept the panel’s recommendation. As part of
the FDA’s review of a PMA, the FDA will typically inspect the manufacturer’s facilities for compliance with QSR
requirements, which impose requirements related to design controls, manufacturing controls, documentation and other
quality assurance procedures. The user fee costs and the length of FDA review time for obtaining PMA approval are
significantly higher than for a 510(k) notification or a de novo classification. PMA applications are subject to an
application fee, which for federal fiscal year 2026 is $579,272 and the small business fee is $144,818.
The FDA will approve the new device for commercial distribution if it determines that the data and information in the PMA
constitute valid scientific evidence and that there is reasonable assurance that the device is safe and effective for its
intended use(s). The FDA may approve a PMA with post-approval conditions intended to ensure the safety and
effectiveness of the device, including, among other things, restrictions on labeling, promotion, sale and distribution, and
collection of long-term follow-up data from patients in the clinical study that supported PMA approval or requirements to
conduct additional clinical studies post-approval. The FDA may condition PMA approval on some form of post-market
surveillance when deemed necessary to protect the public health or to provide additional safety and efficacy data for the
device in a larger population or for a longer period of use. In such cases, the manufacturer might be required to follow
certain patient groups for a number of years and to make periodic reports to the FDA on the clinical status of those
patients. Failure to comply with the conditions of approval can result in material adverse enforcement action, including
withdrawal of the approval.
Certain changes to an approved device, such as changes in manufacturing facilities, methods, or quality control
procedures, or changes in the design performance specifications, which affect the safety or effectiveness of the device,
require submission of a PMA supplement. PMA supplements often require submission of the same type of information as
a PMA, except that the supplement is limited to information needed to support any changes from the device covered by
the original PMA and may not require as extensive clinical data or the convening of an advisory panel. Certain other
changes to an approved device require the submission of a new PMA, such as when the design change causes a
different intended use, mode of operation, and technical basis of operation, or when the design change is so significant
139
that a new generation of the device will be developed, and the data that were submitted with the original PMA are not
applicable for the change in demonstrating a reasonable assurance of safety and effectiveness.
510(k) Notification Pathway
To obtain 510(k) clearance, a manufacturer must submit a premarket notification demonstrating to the FDA’s satisfaction
that the proposed device is “substantially equivalent” to another legally marketed device that itself does not require PMA
approval (a predicate device). A predicate device is a legally marketed device that is not subject to premarket approval,
i.e., a device that was legally marketed prior to May 28, 1976 ("pre-amendments device") and for which a PMA is not
required, a device that has been reclassified from Class III to Class II or I, or a device that was found substantially
equivalent through the 510(k) process. To be “substantially equivalent,” the proposed device must have the same
intended use as the predicate device, and either have the same technological characteristics as the predicate device or
have different technological characteristics and not raise different questions of safety or effectiveness than the
predicate device. FDA then determines whether the device is as safe and effective as the predicate device by reviewing
the scientific methods used to evaluate differences in technological characteristics and performance data. The FDA’s
510(k) clearance process usually takes from three to 12 months, but often takes longer. FDA may require additional
information, including clinical data, to make a determination regarding substantial equivalence. In addition, the FDA
collects user fees for certain medical device submissions and annual fees and for medical device establishments. If the
FDA agrees that the device is substantially equivalent to a lawfully marketed predicate device, it will grant 510(k)
clearance to authorize the device for commercialization. If the FDA determines that the device is “not substantially
equivalent” to a previously cleared device, the device is automatically designated as a Class III device. The device
sponsor must then fulfill more rigorous PMA requirements, or can request a risk-based classification determination for
the device in accordance with the de novo process, which is a route to market for novel medical devices that are low to
moderate risk and are not substantially equivalent to a predicate device, discussed below.
After a device receives 510(k) clearance, any modification that could significantly affect its safety or effectiveness, or
that would constitute a major change or modification in its intended use, will require a new 510(k) clearance or,
depending on the modification, PMA approval. The FDA requires each manufacturer to determine whether the proposed
change requires submission of a 510(k) or a PMA in the first instance, but the FDA can review any such decision and
disagree with a manufacturer’s determination. Many minor modifications are accomplished by a “letter to file” in which
the manufacturer documents the rationale for the change and why a new 510(k) is not required.
However, if the FDA disagrees with a manufacturer’s determination, the FDA can require the manufacturer to cease
marketing and/or request the recall of the modified device until 510(k) marketing clearance or PMA approval is obtained.
Also, in these circumstances, the manufacturer may be subject to significant regulatory fines or penalties.
If no legally marketed predicate can be identified for a new device to enable use of the 510(k) pathway, the device is
automatically classified under the FDCA into Class III, which generally requires PMA approval. However, the FDA can
reclassify or a sponsor can seek de novo classification for a novel device that is low to moderate risk and would
otherwise meet the FDCA standards for a Class I or Class II device, permitting the device to be marketed without PMA
approval.
De Novo Classification
The Food and Drug Administration Modernization Act of 1997 established a route to market for low to moderate risk
medical devices that are automatically placed into Class III due to the absence of a predicate device, called the “Request
for Evaluation of Automatic Class III Designation,” or the de novo classification procedure. This procedure allows a
manufacturer whose novel device is automatically classified into Class III to request down-classification of its medical
device into Class I or Class II on the basis that the device presents low or moderate risk, rather than requiring the
submission and approval of a PMA. Prior to the enactment of the Food and Drug Administration Safety and Innovation
Act of 2012 ("FDASIA"), a medical device could be eligible for de novo classification only if the manufacturer first
submitted a 510(k) premarket notification and received a determination from the FDA that the device was not
substantially equivalent to a legally marketed predicate device. FDASIA streamlined the de novo classification pathway
by permitting manufacturers to request de novo classification directly without first submitting a 510(k) premarket
notification to the FDA and receiving a not substantially equivalent determination. If the manufacturer seeks
reclassification into Class II, the manufacturer must include a draft proposal for special controls that are necessary to
provide a reasonable assurance of the safety and effectiveness of the medical device. In addition, the FDA may reject
the request if it identifies a legally marketed predicate device that would be appropriate for a 510(k) notification,
determines that the device is not low to moderate risk, or that general controls would be inadequate to control the risks
and special controls cannot be developed. After a device receives de novo classification, any modification that could
significantly affect its safety or efficacy, or that would constitute a major change or modification in its intended use, will
require a new 510(k) clearance or, depending on the modification, another de novo request or even PMA approval.
Investigational Device Exemption Process
Clinical trials are almost always required to support a PMA and de novo classification and are sometimes required to
support a 510(k) submission. All clinical investigations of investigational devices to determine safety and effectiveness
must be conducted in accordance with the FDA’s investigational device exemption ("IDE") regulations which govern
investigational device labeling, prohibit promotion of the investigational device, and specify an array of recordkeeping,
reporting and monitoring responsibilities of study sponsors and study investigators. If the device presents a “significant
140
risk” to human health, the FDA requires the device sponsor to submit an IDE application to the FDA, which must become
effective prior to commencing human clinical trials.
A significant risk device is one that presents a potential for serious risk to the health, safety or welfare of a patient and
either is implanted, used in supporting or sustaining human life, substantially important in diagnosing, curing, mitigating
or treating disease or otherwise preventing impairment of human health, or otherwise presents a potential for serious risk
to a subject. An IDE application must be supported by appropriate data, such as animal and laboratory test results,
showing that it is safe to test the device in humans and that the testing protocol is scientifically sound. The IDE will
automatically become effective 30 days after receipt by the FDA, unless the FDA notifies the company that the
investigation may not begin. If the FDA determines that there are deficiencies or other concerns with an IDE for which it
requires modification, the FDA may permit a clinical trial to proceed under a conditional approval.
In addition, the study must be approved by, and conducted under the oversight of, an IRB for each clinical site. The IRB is
responsible for the initial and continuing review of the IDE, and may pose additional requirements for the conduct of the
study. If an IDE application is approved by the FDA and one or more IRBs, human clinical trials may begin at a specific
number of investigational sites with a specific number of patients, as approved by the FDA. If the device presents a non-
significant risk to the patient, a sponsor may begin the clinical trial after obtaining approval for the trial by one or more
IRBs without separate approval from the FDA, but must still follow abbreviated IDE requirements, such as monitoring the
investigation, ensuring that the investigators obtain informed consent, and labeling and record-keeping requirements.
Acceptance of an IDE application for review does not guarantee that the FDA will allow the IDE to become effective and,
if it does become effective, the FDA may or may not determine that the data derived from the trials support the safety
and effectiveness of the device or warrant the continuation of clinical trials. An IDE supplement must be submitted to,
and approved by, the FDA before a sponsor or investigator may make a change to the investigational plan that may
affect its scientific soundness, study plan or the rights, safety or welfare of human subjects.
During a study, the sponsor is required to comply with the applicable FDA requirements, including, for example, trial
monitoring, selecting clinical investigators and providing them with the investigational plan, ensuring IRB review, adverse
event reporting, record keeping, and prohibitions on the promotion of investigational devices or on making safety or
effectiveness claims for them. The clinical investigators in the clinical study are also subject to FDA regulations and must
obtain patient informed consent, rigorously follow the investigational plan and study protocol, control the disposition of
the investigational device, and comply with all reporting and recordkeeping requirements.
Expedited Development and Review Programs for Medical Devices
The FDA has implemented a Breakthrough Devices Program, which is a voluntary program offering manufacturers of
certain devices an opportunity to interact with the FDA more frequently and efficiently as they develop their products
with the goal of expediting commercialization of such products to help patients have more timely access. The program is
available to medical devices that meet certain eligibility criteria, including that the device provides more effective
treatment or diagnosis of life-threatening or irreversibly debilitating diseases or conditions, and constitutes a device (i)
that represents a breakthrough technology, (ii) for which no approved or cleared alternatives exist, (iii) that offer
significant advantages over existing approved or cleared alternatives, or (iv) the availability of which is in the best
interest of patients. Devices granted Breakthrough Device designation are eligible to rely on certain features of the
Breakthrough Device Program, including interactive and timely communications with FDA staff, use of post-market data
collection, when scientifically appropriate, to facilitate expedited and efficient development and review of the device,
opportunities for efficient and flexible clinical study design and priority review of premarket submissions.
Post-market Regulation of Medical Devices
After a device is cleared or approved by the FDA for marketing, numerous and pervasive regulatory requirements
continue to apply. These include:
•establishment registration and device listing with the FDA;
•QSR requirements, which require manufacturers, including third-party manufacturers, to follow stringent design,
testing, control, documentation and other quality assurance procedures during all aspects of the design and
manufacturing process;
•labeling regulations and FDA prohibitions against the promotion of “off-label” uses of cleared or approved products;
•requirements related to promotional activities;
•clearance or approval of product modifications to 510(k)-cleared devices that could significantly affect safety or
effectiveness or that would constitute a major change in intended use of cleared devices, or approval of certain
modifications to PMA-approved devices;
•medical device reporting regulations, which require that a manufacturer report to the FDA if a device it markets may
have caused or contributed to a death or serious injury, or has malfunctioned and the device or a similar device that
it markets would be likely to cause or contribute to a death or serious injury, if the malfunction were to recur;
141
•correction, removal and recall reporting regulations, which require that manufacturers report to the FDA field
corrections and product recalls or removals if undertaken to reduce a risk to health posed by the device or to
remedy a violation of the FDCA that may present a risk to health;
•the FDA’s recall authority, whereby the agency can order device manufacturers to recall from the market a product
that is in violation of governing laws and regulations; and
•post-market surveillance activities and regulations, which apply when deemed by the FDA to be necessary to
protect the public health or to provide additional safety and effectiveness data for the device.
Device manufacturing processes subject to FDA oversight are required to comply with the applicable portions of the
QSR, which cover the methods and the facilities and controls for the design, manufacture, testing, production,
processes, controls, quality assurance, labeling, packaging, distribution, installation and servicing of finished devices
intended for human use. The QSR also requires, among other things, maintenance of a device master file, device history
file, and complaint files. Manufacturers are subject to periodic scheduled or unscheduled inspections by the FDA. A
failure to maintain compliance with the QSR requirements could result in the shut-down of, or restrictions on,
manufacturing operations and the recall or seizure of products. The discovery of previously unknown problems with
products, including unanticipated adverse events or adverse events of increasing severity or frequency, whether
resulting from the use of the device within the scope of its clearance or off-label by a physician in the practice of
medicine, could result in restrictions on the device, including the removal of the product from the market or voluntary or
mandatory device recalls.
The FDA has broad regulatory compliance and enforcement powers. If the FDA determines that a manufacturer has failed
to comply with applicable regulatory requirements, it can take a variety of compliance or enforcement actions, including
the following:
•issuance of warning letters, untitled letters, fines, injunctions, consent decrees and civil penalties;
•requesting or requiring recalls, withdrawals, or administrative detention or seizure of our products;
•imposing operating restrictions or partial suspension or total shutdown of production;
•refusing or delaying requests for 510(k) marketing clearance or PMA approvals of new products or modified
products;
•withdrawing 510(k) clearances or PMA approvals that have already been granted;
•refusal to grant export approvals for our products; or
•criminal prosecution.
Pharmaceutical Coverage, Pricing and Reimbursement
In the U.S. and markets in other countries, patients who are prescribed treatments for their conditions and providers
performing the prescribed services generally rely on third-party payors to reimburse all or part of the associated
healthcare costs. Significant uncertainty exists as to the coverage and reimbursement status of products approved by
the FDA and other government authorities. Thus, even if a product candidate is approved, sales of the product will
depend, in part, on the extent to which third-party payors, including government health programs in the U.S. such as
Medicare and Medicaid, commercial health insurers and managed care organizations, provide coverage, and establish
adequate reimbursement levels for, the product. The process for determining whether a payor will provide coverage for a
product may be separate from the process for setting the price or reimbursement rate that the payor will pay for the
product once coverage is approved. Third-party payors are increasingly challenging the prices charged, examining the
medical necessity, and reviewing the cost-effectiveness of medical products and services and imposing controls to
manage costs. Third-party payors may limit coverage to specific products on an approved list, also known as a
formulary, which might not include all of the approved products for a particular indication.
In order to secure coverage and reimbursement for any product that might be approved for sale, a company may need to
conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and cost-effectiveness of
the product, in addition to the costs required to obtain FDA or other comparable marketing approvals. Nonetheless,
product candidates may not be considered medically necessary or cost effective. A decision by a third-party payor not
to cover a product candidate could reduce physician utilization once the product is approved and have a material
adverse effect on sales, results of operations and financial condition. Additionally, a payor’s decision to provide coverage
for a product does not imply that an adequate reimbursement rate will be approved. Further, one payor’s determination
to provide coverage for a drug product does not assure that other payors will also provide coverage and reimbursement
for the product, and the level of coverage and reimbursement can differ significantly from payor to payor.
Under the Outpatient Prospective Payment System ("OPPS") the costs associated with diagnostic radiopharmaceuticals
have been packaged into the payment for the nuclear medicine tests with which they are used. In July 2024, CMS
recognized that in certain instances the payment amount for the nuclear medicine tests may not adequately account for
the cost of certain specialized diagnostic radiopharmaceuticals, even when those agents may be the most clinically
142
appropriate. Accordingly, CMS proposed to revise its policies so as to pay separately for any diagnostic
radiopharmaceutical with a per day cost greater than $630 and removing such costs from the payment amounts for the
nuclear medicine tests. Any diagnostic radiopharmaceutical with a per-day cost equal to or below that threshold would
continue to be policy-packaged, with costs incorporated into the payment rates for the nuclear medicine tests. On
November 1, 2024, CMS issued a final rule to these Medicare payment rates for hospital outpatient and Ambulatory
Surgical Center services for calendar year, finalizing this proposal effective January 1, 2025.
Healthcare Compliance
In the U.S., biopharmaceutical manufacturers and their products are subject to extensive regulation at the federal and
state level, such as laws intended to prevent fraud and abuse in the healthcare industry. Healthcare providers and third-
party payors play a primary role in the recommendation and prescription of pharmaceutical products that are granted
marketing approval. Arrangements with providers, consultants, third-party payors, and customers are subject to broadly
applicable fraud and abuse, anti-kickback, false claims laws, reporting of payments to healthcare providers and patient
privacy laws and regulations and other healthcare laws and regulations that may constrain our business and/or financial
arrangements. Restrictions under applicable federal and state healthcare laws and regulations, including certain laws and
regulations applicable only if we have marketed products, include the following:
•the federal healthcare program Anti-Kickback Statute, which prohibits, among other things, persons from knowingly
and willfully offering, soliciting, receiving, or providing remuneration, directly or indirectly, to induce either the
referral of an individual for, or the purchase, order, or arranging for or recommending the purchase or order of a
good or service for which payment may be made under federal healthcare programs such as Medicare and
Medicaid. A person or entity does not need to have actual knowledge of the federal Anti-Kickback Statute or
specific intent to violate it in order to have committed a violation;
•federal false claims, false statements, and civil monetary penalties laws prohibiting, among other things, any person
from knowingly presenting, or causing to be presented, a false claim for payment of government funds or knowingly
making, or causing to be made, a false statement to get a false claim paid. In addition, the government may assert
that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a
false or fraudulent claim for purposes of the civil False Claims Act;
•the federal Health Insurance Portability and Accountability Act of 1996 ("HIPAA"), which, in addition to privacy
protections applicable to healthcare providers and other entities, prohibits executing a scheme to defraud any
healthcare benefit program or making false statements relating to healthcare matters. Similar to the federal Anti-
Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to
violate it in order to have committed a violation;
•federal laws that require pharmaceutical manufacturers to report certain calculated product prices to the
government or provide certain discounts or rebates to government authorities or private entities, often as a
condition of reimbursement under government healthcare programs;
•federal Open Payments (or federal “sunshine” law), which requires pharmaceutical and medical device companies to
monitor and report certain financial interactions with certain healthcare providers and teaching hospitals to CMS
within the HHS for re-disclosure to the public, as well as ownership and investment interests held by physicians (as
defined by statute) and their immediate family members;
•federal consumer protection and unfair competition laws, which broadly regulate marketplace activities and
activities that potentially harm consumers;
•analogous state laws and regulations, including: state anti-kickback and false claims laws; state laws requiring
pharmaceutical companies to comply with specific compliance standards, restrict financial interactions between
pharmaceutical companies and healthcare providers or require pharmaceutical companies to report information
related to payments to health care providers or marketing expenditures; and state laws governing privacy, security
and breaches of health information in certain circumstances, many of which differ from each other in significant
ways and often are not preempted by HIPAA, thus complicating compliance efforts; and
•laws and regulations prohibiting bribery and corruption such as the FCPA, which, among other things, prohibits U.S.
companies and their employees and agents from authorizing, promising, offering, or providing, directly or indirectly,
corrupt or improper payments or anything else of value to foreign government officials, employees of public
international organizations or foreign government-owned or affiliated entities, candidates for foreign public office,
and foreign political parties or officials thereof.
Violations of these laws are punishable by criminal and/or civil sanctions, including, in some instances, exclusion from
participation in federal and state health care programs, such as Medicare and Medicaid. Ensuring compliance is time
consuming and costly. Similar healthcare laws and regulations exist in the European Union and other jurisdictions,
including reporting requirements detailing interactions with and payments to healthcare providers and laws governing
the privacy and security of personal information.
143
Healthcare Reform
A primary trend in the U.S. healthcare industry and elsewhere is cost containment. There have been a number of federal
and state proposals during the last few years regarding the pricing of drug products, limiting coverage and
reimbursement for medical products and other changes to the healthcare system in the U.S.
In March 2010, the U.S. Congress enacted the ACA, which, among other things, includes changes to the coverage and
payment for pharmaceutical products under government healthcare programs. Since its enactment, there have been
executive, judicial, and Congressional challenges to certain aspects of the ACA.
Other legislative changes have been proposed and adopted since the ACA was enacted. In August 2011, the Budget
Control Act of 2011, among other things, included aggregate reductions to Medicare payments to providers, which went
into effect in April 2013 and will remain in effect through 2032, with the exception of a temporary suspension from May 1,
2020 through March 31, 2022. In addition, on March 11, 2021, the American Rescue Plan Act of 2021 was signed into law,
which eliminated the statutory cap on the Medicaid drug rebate, beginning January 1, 2024. The rebate was previously
capped at 100% of a drug’s average manufacturer price.
Pursuant to subsequent legislation, these Medicare sequester reductions were suspended and reduced in 2021 and 2022
but, as of July 1, 2022, the full 2% cut has resumed. Under current legislation, the actual reductions in Medicare
payments may vary up to 4%. The Consolidated Appropriations Act, which was signed into law by President Biden in
December 2022, made several changes to sequestration of the Medicare program. Section 1001 of the Act delayed the
4% Statutory Pay-As-You-Go Act of 2010 ("PAYGO") sequester for two years, through the end of calendar year 2024.
Triggered by enactment of the American Rescue Plan Act of 2021, the 4% cut to the Medicare program would have taken
effect in January 2023. The Act’s health care offset title includes Section 4163, which extends the 2% Budget Control Act
of 2011 Medicare sequester for six months into FY 2032 and lowers the payment reduction percentages in FYs 2030 and
2031. On July 4, 2025, President Trump signed into law the 2025 Reconciliation Law (Public Law 119-21, commonly
referred to as the "One Big Beautiful Bill Act"), which is estimated to increase the federal deficit substantially. As a result
of this deficit increase, PAYGO is expected to be triggered, requiring additional mandatory Medicare payment reductions
of up to 4%. There is uncertainty regarding how the Budget Control Act sequestration (2%) and PAYGO sequestration (up
to 4%) would be applied if both are simultaneously in effect. Neither statute explicitly addresses whether the reductions
would be combined (resulting in up to 6% total reduction) or whether there is an overall cap that would limit the
combined reduction.
As an alternative to the Affordable Care Act, President Trump recently announced the Great Healthcare Plan. As
presented, the plan is intended to lower drug prices by increasing competition and benchmarking U.S. drug prices to
other countries, reduce insurance premiums by redirecting subsidies from insurers to individuals, increase accountability
and transparency from insurers, and promote consumer choice by giving individuals more direct control over how
healthcare dollars are spent. Legislative and regulatory action will be required to fully implement the plan. It is unclear
how these proposed changes will impact our business and the pharmaceutical industry in general.
Pharmaceutical Prices
The prices of prescription pharmaceuticals have also been the subject of considerable discussion in the U.S. There have
been several recent U.S. Congressional inquiries, as well as proposed and enacted state and federal legislation designed
to, among other things, bring more transparency to pharmaceutical pricing, review the relationship between pricing and
manufacturer patient programs, and reduce the costs of pharmaceuticals under Medicare and Medicaid.
In October 2020, HHS and the FDA published a final rule allowing states and other entities to develop a Section 804
Importation Program ("SIP") to import certain prescription drugs from Canada into the U.S. Section 804 of the FDCA
allows importation of prescription drugs from Canada to significantly reduce the cost of these drugs to the American
consumer without imposing additional risk to public health and safety. Importation program proposals must be submitted
to FDA for review and authorization. Several states have passed laws allowing for the importation of products from
Canada. Other states have passed legislation establishing workgroups to examine the impact of a state importation
program. In May 2025, FDA announced it will offer individual states the opportunity to submit draft proposals for pre-
review and obtain initial feedback from FDA prior to formally submitting their program proposal. States will have an
opportunity to meet with FDA with the goal of reducing the burden on the states and to help them develop robust
proposals. FDA is also working to assist states with options to streamline the required cost savings analysis, and to
provide input regarding the information the states may rely on to estimate cost savings for American consumers.
Further, on November 20, 2020, HHS finalized a regulation removing safe harbor protection for price reductions from
pharmaceutical manufacturers to plan sponsors under Part D, either directly or through pharmacy benefit managers,
unless the price reduction is required by law. The final rule would also eliminate the current safe harbor for Medicare
drug rebates and create new safe harbors for beneficiary point-of-sale discounts and pharmacy benefit manager service
fees. It was originally set to go into effect on January 1, 2022, but with passage of the IRA, has been delayed by
Congress to January 1, 2032.
On August 16, 2022, the IRA was signed into law by President Biden. The new legislation has implications for Medicare
Part D, which is a program available to individuals who are entitled to Medicare Part A or enrolled in Medicare Part B to
give them the option of paying a monthly premium for outpatient prescription drug coverage. Among other things, the
IRA requires manufacturers of certain drugs to engage in price negotiations with Medicare (beginning in 2026), with
144
prices that can be negotiated subject to a cap; imposes rebates under Medicare Part B and Medicare Part D to penalize
price increases that outpace inflation (which began in 2023); and replaces the Part D coverage gap discount program
with a new discounting program (which began in 2025). The IRA permits the Secretary of the HHS to implement many of
these provisions through guidance, as opposed to regulation, for the initial years.
Specifically, with respect to price negotiations, Congress authorized Medicare to negotiate lower prices for certain costly
single-source drug and biologic products that do not have competing generics or biosimilars and are reimbursed under
Medicare Part B and Part D. CMS may negotiate prices for ten high-cost drugs paid for by Medicare Part D starting in
2026, followed by 15 Part D drugs in 2027, 15 Part B or Part D drugs in 2028, and 20 Part B or Part D drugs in 2029 and
beyond. This provision applies to drug products that have been approved for at least nine years and biologics that have
been licensed for 13 years, but it does not apply to drugs and biologics that have been approved for a single rare disease
or condition. Further, the legislation subjects drug manufacturers to civil monetary penalties and a potential excise tax for
failing to comply with the legislation. The legislation also requires manufacturers to pay rebates for drugs in Medicare
Part D whose price increases exceed inflation. The law also capped Medicare out-of-pocket drug costs at approximately
US$4,000 a year in 2024 and, thereafter beginning in 2025, at US$2,000 a year (indexed for inflation in subsequent
years).
The first cycle of negotiations for the Medicare Drug Price Negotiation Program commenced in the summer of 2023. On
August 15, 2024, the HHS published the negotiated “maximum fair prices” for ten selected Part D drugs that treat a range
of conditions, including diabetes, chronic kidney disease, and rheumatoid arthritis. The prices of these ten drugs will
become effective January 1, 2026. On January 17, 2025, CMS announced its selection of 15 additional Part D drugs for
the second cycle of negotiations. The negotiated prices for this second group will be effective on January 1, 2027. While
there had been some questions about the Trump Administration’s position on this program, on September 30, 2025, CMS
issued final guidance for the third negotiation cycle. On January 27, 2026, CMS announced its selection of 15 additional
drugs (covered under either Part B or Part D) for the third cycle of negotiations. The negotiated prices for this third group
will be effective on January 1, 2028. CMS also selected one previously negotiated drug for the program’s first
renegotiations.
On June 6, 2023, Merck filed a lawsuit against HHS and CMS asserting that, among other things, the IRA’s Drug Price
Negotiation Program for Medicare constitutes an uncompensated taking in violation of the Fifth Amendment of the
Constitution. Subsequently, a number of other parties, also filed lawsuits in various courts with similar constitutional
claims against HHS and CMS. As of October 2025, Merck’s action is pending motions for summary judgment, and the
other lawsuits have been largely unsuccessful for the plaintiffs. Multiple federal district courts have granted summary
judgment in favor of HSS, and several of these decisions have been affirmed by federal appellate courts. For example, in
2025, the U.S. Court of Appeals for the Third Circuit rejected appeals from several manufacturers, including AstraZeneca
and Novartis, upholding the program’s constitutionality. The Second and Sixth Circuits have also affirmed dismissals of
similar challenges. In September 2025, AstraZeneca filed a petition for a writ of certiorari, asking the U.S. Supreme Court
to review the Third Circuit’s decision. Accordingly, while the IRA is being actively implemented, the pending appeal to the
U.S. Supreme Court creates uncertainty regarding the program’s ultimate legal status. We expect that litigation involving
these and other provisions of the IRA will continue with unpredictable and uncertain results.
In May 2025, President Trump issued an executive order implementing the concept of most-favored nation pricing.
Under this order, the Department of Health and Human Services, in coordination with other federal agencies, is directed
to take actions to ensure that the price of prescription drugs paid by federal health insurers, including Medicare and
Medicaid, is in line with the prices paid in comparably developed nations. It is unclear how this policy will impact our
business.
At the state level, individual states are increasingly aggressive in passing legislation and implementing regulations
designed to control pharmaceutical product pricing, including price or patient reimbursement constraints, discounts,
restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases,
designed to encourage importation from other countries and bulk purchasing. A number of states, for example, require
drug manufacturers and other entities in the drug supply chain, including health carriers, pharmacy benefit managers,
and wholesale distributors, to disclose information about pricing of pharmaceuticals. This is increasingly true with
respect to products approved pursuant to the accelerated approval pathway. State Medicaid programs and other payers
are developing strategies and implementing significant coverage barriers, or refusing to cover these products outright,
arguing that accelerated approval drugs have insufficient or limited evidence despite meeting the FDA’s standards for
accelerated approval. In addition, regional healthcare organizations and individual hospitals are increasingly using
bidding procedures to determine what pharmaceutical products and which suppliers will be included in their prescription
pharmaceutical and other healthcare programs. These measures could reduce the ultimate demand for our Commercial
Products and product candidates, once approved, or put pressure on our product pricing. We expect that additional state
and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal
and state governments will pay for healthcare products and services, which could result in reduced demand for our
Commercial Products and product candidates, if approved, or additional pricing pressures.
Regulation by the Nuclear Regulatory Commission of Radionuclides Used for Medical Purposes
The Nuclear Regulatory Commission ("NRC") and the FDA share federal responsibility for the regulation of medical
devices, drugs and biological products that utilize radionuclides. In August 1993, the two agencies established a
Memorandum of Understanding ("MOU") outlining the respective responsibilities of each agency and identifying ways in
which FDA and NRC should coordinate their regulatory actions involving such products.
145
Under the MOU, FDA maintains full responsibility for review and approval of radiopharmaceuticals under the FDCA for
drugs and the PHSA for biologics. Pursuant to its authority under the Atomic Energy Act, the NRC regulates the medical
use of nuclear materials to protect public health and safety and the environment.
In addition to the MOU, the NRC has issued a Medical Use Policy Statement. It provides that the NRC will: (i) continue to
regulate the uses of radionuclides in medicine as necessary to provide for the radiation safety of workers and the general
public; (ii) not intrude into medical judgments affecting patients, except as necessary to provide for the radiation safety
of workers and the general public; (iii) when justified by the risk to patients, regulate the radiation safety of patients
primarily to assure the use of the radionuclides is in accordance with the physician’s directions; and (iv) in developing a
specific regulatory approach, consider industry and professional standards that define acceptable approaches for
achieving radiation safety.
Consistent with the MOU and to implement its Medical Use Policy, the Commission has established policies and
regulations to govern the use, handling and disposal of byproduct materials for medical purposes. Specifically, the
Commission regulates the medical use of byproduct material through licensing, inspection and investigation of medical,
industrial, academic and commercial facilities and authorization of physician users. These regulations are meant to
provide for the radiation safety of workers, the general public, patients, and human research subjects without interfering
with treatment protocols established by the physician. To that end, the rules set out procedures and standards to govern
the issuance of licenses to facilities seeking to use byproduct material for medical purposes. Medical use licenses are
issued by an Agreement State or, in Non-Agreement States, the NRC.
U.S. Data Privacy Laws
There are numerous U.S. federal and state laws and regulations related to the privacy and security of personal
information. In particular, regulations promulgated pursuant to HIPAA establish privacy and security standards that limit
the use and disclosure of individually identifiable health information, or protected health information, and require the
implementation of administrative, physical and technological safeguards to protect the privacy of protected health
information and ensure the confidentiality, integrity and availability of electronic protected health information.
Determining whether protected health information has been handled in compliance with applicable privacy standards and
our contractual obligations can be complex and may be subject to changing interpretation. If a sponsor fails to comply
with applicable privacy laws, including applicable HIPAA privacy and security standards, it could face civil and criminal
penalties. HHS enforcement activity can result in financial liability and reputational harm, and responses to such
enforcement activity can consume significant internal resources. In addition, state attorneys general are authorized to
bring civil actions seeking either injunctions or damages in response to violations that threaten the privacy of state
residents.
In addition to potential enforcement by HHS, a sponsor is also potentially subject to privacy enforcement from the
Federal Trade Commission ("FTC"). The FTC has been particularly focused on the unpermitted processing of health and
genetic data through its recent enforcement actions and is expanding the types of privacy violations that it interprets to
be “unfair” under Section 5 of the FTC Act, as well as the types of activities it views to trigger the Health Breach
Notification Rule (which the FTC also has the authority to enforce). The agency is also in the process of developing rules
related to commercial surveillance and data security. Sponsors will need to account for the FTC’s evolving rules and
guidance for proper privacy and data security practices in order to mitigate risk for a potential enforcement action, which
may be costly.
States are also active in creating specific rules relating to the processing of personal information. In 2018, California
passed into law the CCPA, which took effect on January 1, 2020 and imposed many requirements on businesses that
process the personal information of California residents. Many of the CCPA’s requirements are similar to those found in
the GDPR, which is further described below, including requiring businesses to provide notice to data subjects regarding
the information collected about them and how such information is used and shared, and providing data subjects the right
to request access to such personal information and, in certain cases, request the erasure of such personal information.
The CCPA also affords California residents the right to opt-out of “sales” of their personal information. The CCPA
contains significant penalties for companies that violate its requirements.
In November 2020, California voters passed a ballot initiative for the CPRA, which went into effect on January 1, 2023
and significantly expanded the CCPA to incorporate additional GDPR-like provisions including requiring that the use,
retention and sharing of personal information of California residents be reasonably necessary and proportionate to the
purposes of collection or processing, granting additional protections for sensitive personal information, and requiring
greater disclosures related to notice to residents regarding retention of information. The CPRA also created a new
enforcement agency – the California Privacy Protection Agency – the sole responsibility of which is to enforce the CPRA
and other California privacy laws, which will further increase compliance risk. In addition to California, at least 18 other
states have passed comprehensive privacy laws. These laws may impact our business activities, including our
identification of research subjects, relationships with business partners, and ultimately the marketing and distribution of
our Commercial Products and our product candidates, if approved.
Plaintiffs’ lawyers are also increasingly using privacy-related statutes at both the state and federal level to bring lawsuits
against companies for their data-related practices. In particular, there have been a significant number of cases filed
against companies for their use of pixels and other web trackers. These cases often allege violations of the California
Invasion of Privacy Act and other state laws regulating wiretapping, as well as the federal Video Privacy Protection Act.
146
Review and Approval of Medical Products in the European Union
In addition to regulations in the U.S. there are many other jurisdictions that govern clinical trials and commercial sales and
distribution of our products outside of the U.S. Whether or not we obtain FDA approval for a product candidate, we must
obtain approval by the comparable regulatory authorities of foreign countries or economic areas, such as the 27-member
European Union, before we may commence clinical trials or market products in those countries or areas. In the European
Union, our product candidates also may be subject to extensive regulatory requirements. As in the U.S., medicinal
products can be marketed only if a marketing authorization from the competent regulatory agencies has been obtained.
Similar to the U.S., the various phases of preclinical and clinical research in the European Union are subject to significant
regulatory controls.
The approval process and requirements governing the conduct of clinical trials, product licensing, pricing, and
reimbursement can vary from country to country and can involve additional testing and additional administrative review
periods. The time required to obtain approval in other countries might differ from and be longer than that required to
obtain FDA approval. Regulatory approval in one country or jurisdiction does not ensure regulatory approval in another,
but a failure or delay in obtaining regulatory approval in one country or jurisdiction may negatively impact the regulatory
process in others.
Non-clinical Studies
Non-clinical studies are performed to demonstrate the health or environmental safety of new chemical or biological
substances. Non-clinical (pharmaco-toxicological) studies must be conducted in compliance with the principles of GLP,
as set forth in EU Directive 2004/10/EC (unless otherwise justified for certain particular medicinal products – e.g., radio-
pharmaceutical precursors for radio-labeling purposes). In particular, non-clinical studies, both in vitro and in vivo, must
be planned, performed, monitored, recorded, reported and archived in accordance with the GLP principles, which define
a set of rules and criteria for a quality system for the organizational process and the conditions for non-clinical studies.
These GLP standards reflect the Organization for Economic Co-operation and Development requirements.
Clinical Studies
Clinical trials of medicinal products in the European Union must be conducted in accordance with EU and national
regulations and the ICH guidelines on GCP, as well as the applicable regulatory requirements and the ethical principles
that have their origin in the Declaration of Helsinki. If the sponsor of the clinical trial is not established within the
European Union, it must appoint an EU entity to act as its legal representative. The sponsor must take out a clinical trial
insurance policy, and in most EU member states, the sponsor is liable to provide ‘no fault’ compensation to any study
subject injured in the clinical trial.
The regulatory landscape related to clinical trials in the European Union has been subject to recent changes. On January
31, 2022, the new Clinical Trials Regulation (EU) No 536/2014 became applicable in the European Union and repealed
and replaced the prior Clinical Trials Directive 2001/20/EC. Unlike directives, the new Regulation is directly applicable in
all EU member states without the need for member states to further implement it into national law. It aims at simplifying
and streamlining the authorization, conduct and transparency of clinical trials in the European Union.
Under the new coordinated procedure, the sponsor of a clinical trial to be conducted in more than one member state will
only be required to submit a single application. The Regulation allows sponsors to make a single submission to both the
competent authority and an ethics committee in each member state, leading to a single decision per member state. The
submission will be made through the Clinical Trials Information System, a new clinical trials portal overseen by the EMA
and available to clinical trial sponsors, competent authorities of the EU member states and the public.
Beyond streamlining the process, the new Regulation includes a single set of documents to be prepared and submitted
for the application and a harmonized procedure for the assessment of applications for clinical trials, which is divided in
two parts. Part I is subject to a coordinated review by competent authorities of all EU member states in which an
application for authorization has been submitted ("member states concerned"). One of the member states concerned
("the reporting member state") prepares a draft assessment report which is submitted to other member states concerned
for their joint review, allowing for a single assessment report to be issued at the term of the assessment process. Part II
is assessed separately by each member state concerned. Strict deadlines have been established for the assessment of
clinical trial applications ("CTAs"). The role of the relevant ethics committees in the assessment procedure will continue
to be governed at national levels; however, overall related timelines are set out under the Clinical Trials Regulation. The
Regulation also provides for simplified reporting procedures for clinical trial sponsors.
All ongoing clinical trials in the European Union approved under the prior Clinical Trials Directive must be transitioned to
the Clinical Trials Information System by January 31, 2025. This date marks the end of a three-year transition period that
began when the Clinical Trials Regulation became applicable in the European Union on January 31, 2022. Clinical trials
that were started under the Clinical Trials Directive and are subject to transition to the Clinical Trials Regulation will by
January 31, 2025 have to comply with the obligations of the Clinical Trials Regulation even if these are not included in the
previous study protocol, such as (i) obligations of notification via the Clinical Trials Information System; (ii) safety
reporting rules; (iii) archiving requirement; and (iv) transparency requirements. Failure to transition ongoing clinical trials
to the Clinical Trials Regulation by January 31, 2025 can result in corrective measures under Article 77 of the Clinical
Trials Regulation, including revocation of the authorization of the clinical trial or suspension of the clinical trial, as well as
criminal sanctions and fines under national law of EU Member States.
147
Parties conducting certain clinical trials must, as in the U.S., post clinical trial information in the European Union at the
EudraCT website.
Marketing Authorization
In order to market our product candidates in the European Union and many other foreign jurisdictions, we must obtain
separate regulatory approvals. More concretely, in the European Union, medicinal product candidates can only be
commercialized after obtaining a marketing authorization ("MA"). To obtain regulatory approval of a product candidate
under EU regulatory systems, we must submit a MA application ("MAA"). The process for doing this depends, among
other things, on the nature of the medicinal product. There are two types of MAs: “Centralized MAs” are issued by the
European Commission through the centralized procedure based on the opinion of the Committee for Medicinal Products
for Human Use ("CHMP") of the EMA and are valid throughout the European Union. The centralized procedure is
compulsory for certain types of medicines such as (i) medicinal products developed by specified biotechnological
processes, (ii) products designated orphan medicinal products, (iii) advanced-therapy medicines (such as gene-therapy,
somatic cell-therapy or tissue-engineered medicines), and (iv) products with a new active substance indicated for the
treatment of specified diseases, such as HIV/AIDS, cancer, diabetes, neurodegenerative disorders or autoimmune
diseases, and other immune dysfunctions and viral diseases. The centralized procedure is optional for products
containing a new active substance not yet authorized in the European Union, or for products that represent a significant
therapeutic, scientific, or technical innovation, or whose authorization would be in the interest of public health.
Under the centralized procedure the maximum timeframe for the evaluation of an MAA by the EMA is 210 days, excluding
clock stops, when additional written or oral information is to be provided by the sponsor in response to questions asked
by the CHMP. Accelerated assessment might be granted by the CHMP in exceptional cases, when a medicinal product is
expected to be of a major public health interest, particularly from the point of view of therapeutic innovation. The
timeframe for the evaluation of an MAA under the accelerated assessment procedure is 150 days, excluding stop-clocks.
“National MAs” are issued by the competent authorities of the EU member states, only cover their respective territory,
and are available for product candidates not falling within the mandatory scope of the centralized procedure. Under the
mutual recognition procedure, a medicine is first authorized in one EU member state (which acts as the reference
member state), in accordance with the national procedures of that member state. Following this, further MAs can be
progressively sought from other EU member states in a procedure whereby the member states concerned agree to
recognize the validity of the original, national MA produced by the reference member state. Under the decentralized
procedure, if the product has not received a national MA in any member state at the time of application, a sponsor may
apply for simultaneous authorization in more than one EU member state. Under the decentralized procedure an identical
dossier is submitted to the competent authorities of each of the member states in which the MA is sought, one of which
is selected by the applicant as the reference member state.
Conditional Marketing Authorization
In particular circumstances, a “conditional” MA may be granted in cases where all the required safety and efficacy data
are not yet available. A conditional MA is subject to conditions to be fulfilled for generating the missing data or ensuring
increased safety measures. Conditional MAs are valid for one year, and may be renewed annually, if the risk-benefit
balance remains positive, and after an assessment of the need for additional or modified conditions or specific
obligations. Once the pending studies are provided, it can become a “standard” MA. However, if the conditions are not
fulfilled within the timeframe set by the EMA, the MA ceases to be renewed. The timelines for the centralized procedure
described above also apply with respect to the review by the CHMP of applications for a conditional MA, but sponsors
can also request the EMA to conduct an accelerated assessment, for instance in cases of unmet medical needs.
Marketing Authorization Granted under Exceptional Circumstances
A MA may also be granted “under exceptional circumstances” when the applicant can show that it is unable to provide
comprehensive data on the efficacy and safety under normal conditions of use even after the product has been
authorized and subject to specific procedures being introduced. This may arise in particular when the intended
indications are very rare and, in the present state of scientific knowledge, it is not possible to provide comprehensive
information, or when generating data may be contrary to generally accepted ethical principles. This MA is close to the
conditional MA as it is reserved to medicinal products to be approved for severe diseases or unmet medical needs and
the applicant does not hold the complete data set legally required for the grant of a MA. However, unlike the conditional
MA, the applicant does not have to provide the missing data and will never have to. Although the MA “under exceptional
circumstances” is granted definitively, the risk-benefit balance of the medicinal product is reviewed annually and the MA
is withdrawn in case the risk-benefit ratio is no longer favorable.
Under the above-described procedures, before granting the MA, the EMA or the competent authorities of the member
states make an assessment of the risk-benefit balance of the product on the basis of scientific criteria concerning its
quality, safety, and efficacy. Except conditional MAs, MAs have an initial duration of five years. After these five years, the
authorization may be renewed on the basis of a reevaluation of the risk-benefit balance.
Pediatric Studies
Prior to obtaining a marketing authorization in the European Union, sponsors have to demonstrate compliance with all
measures included in an EMA-approved Pediatric Investigation Plan ("PIP") covering all subsets of the pediatric
population, unless the EMA has granted a product-specific waiver, a class waiver, or a deferral for one or more of the
148
measures included in the PIP. The respective requirements for all marketing authorization procedures are set forth in
Regulation (EC) No 1901/2006, which is referred to as the Pediatric Regulation. This requirement also applies when a
company wants to add a new indication, pharmaceutical form, or route of administration for a medicine that is already
authorized. The Pediatric Committee of the EMA ("PDCO") may grant deferrals for some medicines, allowing a company
to delay development of the medicine in children until there is enough information to demonstrate its effectiveness and
safety in adults. The PDCO may also grant waivers when development of a medicine in children is not needed or is not
appropriate because (i) the product is likely to be ineffective or unsafe in part or all of the pediatric population; (ii) the
disease or condition occurs only in adult population; or (iii) the product does not represent a significant therapeutic
benefit over existing treatments for pediatric population. Before a marketing authorization application can be filed, or an
existing marketing authorization can be amended, the EMA determines that companies actually comply with the agreed
studies and measures listed in each relevant PIP.
PRIME Designation
In March 2016, the EMA launched an initiative to facilitate development of product candidates in indications, often rare,
for which few or no therapies currently exist. The Priority Medicines ("PRIME") scheme is intended to encourage drug
development in areas of unmet medical need and provides accelerated assessment of products representing substantial
innovation reviewed under the centralized procedure. Products from small- and medium-sized enterprises ("SMEs") may
qualify for earlier entry into the PRIME scheme than larger companies. Many benefits accrue to sponsors of product
candidates with PRIME designation, including but not limited to, early and proactive regulatory dialogue with the EMA,
frequent discussions on clinical trial designs and other development program elements, and accelerated MAA
assessment once a dossier has been submitted. Importantly, a dedicated agency contact and rapporteur from the CHMP
or Committee for Advanced Therapies are appointed early in the PRIME scheme, facilitating increased understanding of
the product at EMA’s Committee level. A kick-off meeting initiates these relationships and includes a team of
multidisciplinary experts at the EMA to provide guidance to the sponsor on the overall development and regulatory
strategies.
Periods of Authorization and Renewals
A marketing authorization is valid for five years in principle and the marketing authorization may be renewed after five
years on the basis of a re-evaluation of the risk-benefit balance by the EMA or by the competent authority of the
authorizing member state. To this end, the marketing authorization holder must provide the EMA or the competent
authority with a consolidated version of the file in respect of quality, safety, and efficacy, including all variations
introduced since the marketing authorization was granted, at least nine months before the marketing authorization
ceases to be valid. Once renewed, the marketing authorization is valid for an unlimited period, unless the European
Commission or the competent authority decides, on justified grounds relating to pharmacovigilance, to proceed with one
additional five-year renewal. Any authorization which is not followed by the actual placing of the drug on the EU market
(in case of centralized procedure) or on the market of the authorizing member state within three years after authorization
ceases to be valid (the so-called "sunset clause").
Regulatory Exclusivity
In the European Union, new products authorized for marketing (i.e., reference products) qualify for eight years of data
exclusivity and an additional two years of market exclusivity upon marketing authorization. The data exclusivity period
prevents generic sponsors from relying on the preclinical and clinical trial data contained in the dossier of the reference
product when applying for a generic marketing authorization in the European Union during a period of eight years from
the date on which the reference product was first authorized in the European Union. The market exclusivity period
prevents a successful generic sponsor from commercializing its product in the European Union until ten years have
elapsed from the initial authorization of the reference product in the European Union. The ten-year market exclusivity
period can be extended to a maximum of 11 years if, during the first eight years of those ten years, the marketing
authorization holder obtains an authorization for one or more new therapeutic indications which, during the scientific
evaluation prior to their authorization, are held to bring a significant clinical benefit in comparison with existing therapies.
There is a special regime for biosimilars, or biological medicinal products that are similar to a reference medicinal product
but that do not meet the definition of a generic medicinal product, for example, because of differences in raw materials
or manufacturing processes. For such products, the results of appropriate preclinical or clinical trials must be provided,
and guidelines from the EMA detail the type of quantity of supplementary data to be provided for different types of
biological product. There are no such guidelines for complex biological products, such as gene or cell therapy medicinal
products, and so it is unlikely that biosimilars of those products will currently be approved in the European Union.
In this context, it should be noted that the EU pharmaceutical legislation is currently undergoing a complete review
process, in the context of the Pharmaceutical Strategy for Europe initiative, launched by the European Commission in
November 2020. The European Commission’s proposal for revision of several legislative instruments related to medicinal
products was published in April 2023 and includes, among other things, provisions that would potentially reduce the
duration of regulatory data protection. The European Parliament requested several amendments in April 2024. At this
time, the proposed revisions remain to be agreed and adopted by the European Parliament and European Council and the
proposals may therefore be substantially revised before adoption, which is not anticipated before early 2026. The
revisions may, however, have a significant impact on the pharmaceutical industry in the long term, if and when adopted.
149
However, guidance from the EMA states that they will be considered in the future in light of the scientific knowledge and
regulatory experience gained at the time.
Orphan Drug Designation and Exclusivity
The criteria for designating an orphan medicinal product in the European Union are similar in principle to those in the
United States. Under Article 3 of Regulation (EC) 141/2000, a medicinal product may be designated as orphan if (i) it is
intended for the diagnosis, prevention, or treatment of a life-threatening or chronically debilitating condition, (ii) either (a)
such condition affects no more than five in 10,000 persons in the European Union when the application is made, or (b)
the product, without the benefits derived from orphan status, would not generate sufficient return in the European Union
to justify investment, and (iii) there exists no satisfactory method of diagnosis, prevention or treatment of such condition
authorized for marketing in the European Union, or if such a method exists, the product will be of significant benefit to
those affected by the condition. The term ‘significant benefit’ is defined in Regulation (EC) 847/2000 to mean a clinically
relevant advantage or a major contribution to patient care.
Orphan medicinal products are eligible for financial incentives such as reduction of fees or fee waivers and are, upon
grant of a marketing authorization, entitled to ten years of market exclusivity for the approved therapeutic indication.
During this ten year market exclusivity period, the EMA or the competent authorities of the Member States of the
European Economic Area ("EEA") cannot accept an application for a marketing authorization for a similar medicinal
product for the same indication. A similar medicinal product is defined as a medicinal product containing a similar active
substance or substances as contained in an authorized orphan medicinal product, and which is intended for the same
therapeutic indication. The application for orphan designation must be submitted before the application for marketing
authorization. The sponsor will receive a fee reduction for the MAA if the orphan designation has been granted, but not if
the designation is still pending at the time the marketing authorization is submitted. Orphan designation does not convey
any advantage in, or shorten the duration of, the regulatory review and approval process.
The ten-year market exclusivity in the European Union may be reduced to six years if, at the end of the fifth year, it is
established that the product no longer meets the criteria for orphan designation, for example, if the product is sufficiently
profitable not to justify maintenance of market exclusivity. Additionally, marketing authorization may be granted to a
similar product for the same indication at any time if: (i) the second sponsor can establish that its product, although
similar, is safer, more effective, or otherwise clinically superior; (ii) the sponsor consents to a second orphan medicinal
product application; or (iii) the sponsor cannot supply enough orphan medicinal product.
Pediatric Exclusivity
If a sponsor obtains a marketing authorization in all EU Member States, or a marketing authorization granted in the
centralized procedure by the European Commission, and the study results for the pediatric population are included in the
product information, even when negative, the medicine is then eligible for an additional six-month period of qualifying
patent protection through extension of the term of the Supplementary Protection Certificate ("SPC") or alternatively a
one year extension of the regulatory market exclusivity from ten to eleven years, as selected by the marketing
authorization holder.
Post-Approval Requirements
As in the U.S., both MA holders and manufacturers of medicinal products are subject to comprehensive regulatory
oversight by the EMA, the European Commission, and the competent authorities of EU member states. The MA holder
must, for example, comply with EU pharmacovigilance legislation and its related regulations and guidelines which entail
many requirements for conducting pharmacovigilance, or the assessment and monitoring of the safety of medicinal
products. In particular, the MA holder must establish and maintain a pharmacovigilance system and appoint an individual
qualified person for pharmacovigilance ("QPPV") who is responsible for the establishment and maintenance of that
system, and oversees the safety profiles of medicinal products and any emerging safety concerns. Key obligations
include expedited reporting of suspected serious adverse reactions and submission of periodic safety update reports
("PSURs").
All new MAs must include a risk management plan ("RMP"), describing the risk management system that the company
will put in place and documenting measures to prevent or minimize the risks associated with the product. The regulatory
authorities may also impose specific obligations as a condition of the MA. Such risk-minimization measures or post-
authorization obligations may include additional safety monitoring, more frequent submission of PSURs, or the conduct of
additional clinical trials or post-authorization safety studies.
The manufacturing process for medicinal products in the European Union is also highly regulated and regulators may
shut down manufacturing facilities that they believe do not comply with regulations. Manufacturing requires a
manufacturing authorization, and the manufacturing authorization holder must comply with various requirements set out
in the applicable EU laws, including compliance with EU GMP standards when manufacturing medicinal products and API.
In the European Union, the advertising and promotion of approved products are subject to laws governing promotion of
medicinal products, interactions with physicians, misleading and comparative advertising, and unfair commercial
practices. These laws require that promotional materials and advertising in relation to medicinal products comply with the
product’s Summary of Product Characteristics ("SmPC") as approved by the competent authorities. Promotion of a
medicinal product that does not comply with the SmPC is considered to constitute off-label promotion, which is
150
prohibited in the European Union. Direct-to-consumer advertising of prescription medicines is also prohibited in the
European Union. Although general requirements for advertising and promotion of medicinal products are established
under EU directives, the details are governed by regulations in each member state and can differ from one country to
another.
The aforementioned EU rules are generally applicable in the EEA.
Failure to comply with European Union and member state laws that apply to the conduct of clinical trials, manufacturing
approval, MA of medicinal products and marketing of such products, both before and after grant of the MA,
manufacturing of pharmaceutical products, statutory health insurance, bribery and anti-corruption or with other
applicable regulatory requirements may result in administrative, civil or criminal penalties. These penalties could include
delays or refusal to authorize the conduct of clinical trials, or to grant MA, product withdrawals and recalls, product
seizures, suspension, withdrawal or variation of the MA, total or partial suspension of production, distribution,
manufacturing or clinical trials, operating restrictions, injunctions, suspension of licenses, fines and criminal penalties.
Supplementary Protection Certificate
The European Union also provides for patent term extension through SPCs. The rules and requirements for obtaining a
SPC are similar to those in the U.S. An SPC may extend the term of a patent for up to five years after its originally
scheduled expiration date and can provide up to a maximum of 15 years of marketing exclusivity for a drug. In certain
circumstances, these periods may be extended for six additional months (see “Pediatric Development”).
Although SPCs are available throughout the European Union, sponsors must apply on a country-by-country basis. Similar
patent term extension rights exist in certain other foreign jurisdictions outside the European Union.
Reimbursement and Pricing of Prescription Pharmaceuticals
In international markets including the European Union, reimbursement and healthcare payment systems vary significantly
by country, and many countries have instituted price ceilings on specific products and therapies. In the European Union,
pricing and reimbursement schemes vary widely from country to country. Some countries may require the completion of
additional studies that compare the cost-effectiveness of a particular medicinal product candidate to currently available
therapies. This Health Technology Assessment ("HTA") process, which is currently governed by the national laws of the
individual EU member states, is the procedure according to which the assessment of the public health impact,
therapeutic impact and the economic and societal impact of use of a given medicinal product in the national healthcare
systems of the individual country is conducted. The outcome of HTA regarding specific medicinal products will often
influence the pricing and reimbursement status granted to these medicinal products by the competent authorities of
individual EU member states.
The downward pressure on healthcare costs in general, particularly prescription medicines, has become very intense.
Pharmaceutical products may face competition from lower-priced products in foreign countries that have placed price
controls on pharmaceutical products and may also compete with imported foreign products. Furthermore, there is no
assurance that a product will be considered medically reasonable and necessary for a specific indication, will be
considered cost-effective by third-party payors, that an adequate level of reimbursement will be established even if
coverage is available or that the third-party payors’ reimbursement policies will not adversely affect the ability for
manufacturers to sell products profitably. Historically, products launched in the European Union do not follow price
structures of the U.S. and generally prices tend to be significantly lower.
Healthcare Reform
In the European Union, similar political, economic, and regulatory developments to those in the U.S. may affect our ability
to profitably commercialize our product candidates, if approved. In many countries, including those of the European
Union, the pricing of prescription pharmaceuticals is subject to governmental control and access. In these countries,
pricing negotiations with governmental authorities can take considerable time after the receipt of a marketing approval
for a product. To obtain reimbursement or pricing approval in some countries, pharmaceutical firms may be required to
conduct a clinical trial that compares the cost-effectiveness of the product to other available therapies. In addition to
continuing pressure on prices and cost containment measures, legislative developments at the EU or member state level
may result in significant additional requirements or obstacles. The delivery of healthcare in the European Union, including
the establishment and operation of health services and the pricing and reimbursement of medicines, is almost exclusively
a matter for national, rather than EU, law and policy. National governments and health service providers have different
priorities and approaches to the delivery of health care and the pricing and reimbursement of products in that context. In
general, however, the healthcare budgetary constraints in most EU member states have resulted in restrictions on the
pricing and reimbursement of medicines by relevant health service providers. Coupled with ever-increasing EU and
national regulatory burdens on those wishing to develop and market products, this could restrict or regulate post-
approval activities and affect the ability of pharmaceutical companies to commercialize their products. In international
markets, reimbursement and healthcare payment systems vary significantly by country, and many countries have
instituted price ceilings on specific products and therapies.
In the European Union, potential reductions in prices and changes in reimbursement levels could be the result of different
factors, including reference pricing used by various EU member states, and parallel distribution and parallel trade can
further reduce prices. It could also result from the application of external reference pricing mechanisms, which consist of
151
arbitrage between low-priced and high-priced member states). There can be no assurance that any country that has
price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing
arrangements for any product candidates, if approved in those countries.
HTA of medicinal products in the European Union is an essential element of the pricing and reimbursement decision-
making process in a number of EU member states. The outcome of HTA has a direct impact on the pricing and
reimbursement status granted to the medicinal product. A negative HTA by a leading and recognized HTA body
concerning a medicinal product could undermine the prospects to obtain reimbursement for such product not only in the
EU member state in which the negative assessment was issued, but also in other EU member states.
In 2011, Directive 2011/24/EU was adopted at the EU level. This Directive establishes a voluntary network of national
authorities or bodies responsible for HTA in the individual EU member states. The network facilitates and supports the
exchange of scientific information concerning HTAs. Further to this, on December 13, 2021, Regulation No 2021/2282 on
HTA, amending Directive 2011/24/EU, was adopted. While the Regulation entered into force in January 2022, it will only
begin to apply from January 2025 onwards, with preparatory and implementation-related steps to take place in the
interim. Once applicable, it will have a phased implementation depending on the concerned products. The Regulation
intends to boost cooperation among EU member states in assessing health technologies, including new medicinal
products as well as certain high-risk medical devices, and provide the basis for cooperation at the EU level for joint
clinical assessments in these areas. It will permit EU member states to use common HTA tools, methodologies, and
procedures across the European Union, working together in four main areas, including joint clinical assessment of the
innovative health technologies with the highest potential impact for patients, joint scientific consultations whereby
developers can seek advice from HTA authorities, identification of emerging health technologies to identify promising
technologies early, and continuing voluntary cooperation in other areas.
Individual EU member states will continue to be responsible for assessing non-clinical (e.g., economic, social, ethical)
aspects of health technology, and making decisions on pricing and reimbursement.
Regulation of Medical Devices in the European Union
The European Union has adopted specific directives and regulations regulating the design, manufacture, clinical
investigation, conformity assessment, labeling and adverse event reporting for medical devices. Until May 25, 2021,
medical devices were regulated by the EU Medical Devices Directive ("MDD") which has been repealed and replaced by
the EU Medical Devices Regulation ("MDR"). However, as of May 26, 2021, some of the MDR requirements apply in place
of the corresponding requirements of the MDD with regard to registration of economic operators and of devices, post-
market surveillance and vigilance requirements.
Medical Devices Directive
Under the MDD, all medical devices placed on the market in the European Union must meet the essential requirements
laid down in Annex I to the MDD, including the requirement that a medical device must be designed and manufactured in
such a way that it will not compromise the clinical condition or safety of patients, or the safety and health of users and
others. In addition, the device must achieve the performance intended by the manufacturer and be designed,
manufactured, and packaged in a suitable manner. The European Commission has adopted various standards applicable
to medical devices. These include standards governing common requirements, such as sterilization and safety of medical
electrical equipment and product standards for certain types of medical devices. There are also harmonized standards
relating to design and manufacture. While not mandatory, compliance with these standards and the aforementioned EU
rules is generally applicable in the EEA.
Medical Devices Regulation
The regulatory landscape related to medical devices in the European Union recently evolved. On April 5, 2017, the MDR
was adopted with the aim of ensuring better protection of public health and patient safety. The MDR establishes a
uniform, transparent, predictable and sustainable regulatory framework across the European Union for medical devices
and ensure a high level of safety and health while supporting innovation. Unlike the MDD, the MDR is directly applicable
in EU member states without the need for member states to implement into national law. This aims at increasing
harmonization across the European Union.
The MDR became effective on May 26, 2021. In accordance with its recently extended transitional provisions, both (i)
devices lawfully placed on the market pursuant to the MDD prior to May 26, 2021 and (ii) legacy devices lawfully placed
on the EU market after May 26, 2021 in accordance with the MDR transitional provisions may generally continue to be
made available on the market or put into service, provided that the requirements of the transitional provisions are
fulfilled. However, even in this case, manufacturers must comply with a number of new or reinforced requirements set
forth in the MDR, in particular the obligations described below.
The MDR requires that before placing a device, other than a custom-made device, on the market, manufacturers (as well
as other economic operators such as authorized representatives and importers) must register by submitting identification
information to the electronic system ("Eudamed"), unless they have already registered. The information to be submitted
by manufacturers (and authorized representatives) also includes the name, address and contact details of the person or
persons responsible for regulatory compliance. The new Regulation also requires that before placing a device, other than
a custom-made device, on the market, manufacturers must assign a unique identifier to the device and provide it along
152
with other core data to the unique device identifier ("UDI") database. These new requirements aim at ensuring better
identification and traceability of the devices. Each device – and as applicable, each package – will have a UDI composed
of two parts: a device identifier ("UDI-DI") specific to a device, and a production identifier ("UDI-PI") to identify the unit
producing the device. Manufacturers are also notably responsible for entering the necessary data on Eudamed, which
includes the UDI database, and for keeping it up to date.
All manufacturers placing medical devices on the market in the European Union must comply with the EU medical device
vigilance system which has been reinforced by the MDR. Under this system, serious incidents and Field Safety Corrective
Actions ("FSCAs") must be reported to the relevant authorities of the EU member states. These reports will have to be
submitted through Eudamed – once functional – and aim to ensure that, in addition to reporting to the relevant authorities
of the EU member states, other actors such as the economic operators in the supply chain will also be informed. Until
Eudamed is fully functional, the corresponding provisions of the MDD continue to apply. Manufacturers are required to
take FSCAs, which are defined as any corrective action for technical or medical reasons to prevent or reduce a risk of a
serious incident associated with the use of a medical device that is made available on the market. A serious incident is
any malfunction or deterioration in the characteristics or performance of a device on the market (e.g., inadequacy in the
information supplied by the manufacturer, undesirable side-effect), which, directly or indirectly, might lead to either the
death or serious deterioration of the health of a patient, user, or other persons, or to a serious public health threat.
An FSCA may include the recall, modification, exchange, destruction or retrofitting of the device. FSCAs must be
communicated by the manufacturer or its legal representative to its customers and/or to the end users of the device
through Field Safety Notices. For similar serious incidents that occur with the same device or device type and for which
the root cause has been identified or a FSCA implemented or where the incidents are common and well documented,
manufacturers may provide periodic summary reports instead of individual serious incident reports.
The advertising and promotion of medical devices are subject to some general principles set forth in EU legislation.
According to the MDR, only devices that are CE marked may be marketed and advertised in the European Union in
accordance with their intended purpose. Directive 2006/114/EC concerning misleading and comparative advertising and
Directive 2005/29/EC on unfair commercial practices, while not specific to the advertising of medical devices, also apply
to the advertising thereof and contain general rules, for example, requiring that advertisements are evidenced, balanced
and not misleading. Specific requirements are defined at a national level. EU member states’ laws related to the
advertising and promotion of medical devices, which vary between jurisdictions, may limit or restrict the advertising and
promotion of products to the general public and may impose limitations on promotional activities with healthcare
professionals.
Many EU member states have adopted specific anti-gift statutes that further limit commercial practices for medical
devices, in particular vis-à-vis healthcare professionals and organizations. Additionally, there has been a recent trend of
increased regulation of payments and transfers of value provided to healthcare professionals or entities and many EU
member states have adopted national “Sunshine Acts” which impose reporting and transparency requirements (often on
an annual basis), similar to the requirements in the U.S., on medical device manufacturers. Certain countries also
mandate implementation of commercial compliance programs.
In the European Union, regulatory authorities have the power to carry out announced and, if necessary, unannounced
inspections of companies, as well as suppliers and/or sub-contractors and, where necessary, the facilities of professional
users. Failure to comply with regulatory requirements (as applicable) could require time and resources to respond to the
regulatory authorities’ observations and to implement corrective and preventive actions, as appropriate. Regulatory
authorities have broad compliance and enforcement powers and if such issues cannot be resolved to their satisfaction
can take a variety of actions, including untitled or warning letters, fines, consent decrees, injunctions, or civil or criminal
penalties.
The aforementioned EU rules are generally applicable in the EEA.
EU General Data Protection Regulation
The collection, use, disclosure, transfer, or other processing of personal data regarding individuals in the EEA, including
personal health data, is subject to the GDPR, which became effective on May 25, 2018. In the United Kingdom, the GDPR
is retained in domestic law as the U.K. GDPR and sits alongside an amended version of the UK Data Protection Act. The
GDPR is wide-ranging in scope and imposes numerous requirements on companies that process personal data, including
requirements relating to processing health and other sensitive data, obtaining consent of the individuals to whom the
personal data relates, providing information to individuals regarding data processing activities, implementing safeguards
to protect the security and confidentiality of personal data, providing notification of data breaches, and taking certain
measures when engaging third-party processors. The GDPR also imposes strict rules on the transfer of personal data to
countries outside the European Union, including the U.S., and permits data protection authorities to impose large
penalties for violations of the GDPR, including potential fines of up to €20 million or 4% of annual global revenues of the
respective group of companies, whichever is greater.
The GDPR also confers a private right of action on data subjects and consumer associations to lodge complaints with
supervisory authorities, seek judicial remedies, and obtain compensation for damages resulting from violations of the
GDPR. Compliance with the GDPR is a rigorous and time-intensive process that may increase the cost of doing business
or require companies to change their business practices to ensure full compliance.
153
For transfers of personal data from the European Union to the U.S., the European Commission has adopted an adequacy
decision for the EU-US Data Privacy Framework in July 2023. It is widely expected that this adequacy decision will be
challenged in court, so uncertainties around this issue continue.
Brexit and the Regulatory Framework in the United Kingdom
The United Kingdom’s withdrawal from the European Union, commonly referred to as Brexit, took place on January 31,
2020. The European Union and the United Kingdom reached an agreement on their new partnership in the Trade and
Cooperation Agreement, which entered into force on May 1, 2021. As of January 1, 2021, the MHRA became responsible
for supervising medicines and medical devices in Great Britain, comprising England, Scotland and Wales under domestic
law, whereas Northern Ireland continues to be subject to EU rules under the Northern Ireland Protocol, as amended by
the so called Windsor Framework agreed in February 2023. As of January 1, 2025, the changes introduced by the
Windsor Framework resulted in the MHRA being responsible for approving all medicinal products destined for the United
Kingdom market (Great Britain and Northern Ireland), and the EMA will no longer have any role in approving medicinal
products destined for Northern Ireland. The MHRA relies on the Human Medicines Regulations 2012 (SI 2012/1916), as
amended ("HMR") as the basis for regulating medicines. The HMR has incorporated into the domestic law the body of EU
law instruments governing medicinal products that pre-existed prior to the United Kingdom’s withdrawal from the
European Union.
As of January 1, 2024, a new IRP applies which intends to facilitate approval of pharmaceutical products in the United
Kingdom. The IRP is open to applicants that have already received an authorization for the same product from one of the
MHRA’s specified RRs. The RRs notably include EMA and regulators in the EEA member states for approvals in the EU
centralized procedure and mutual recognition procedure as well as the FDA (for product approvals granted in the U.S.).
The RR assessment must have undergone a full and standalone review. RR assessments based on reliance or recognition
cannot be used to support an IRP application. A CHMP positive opinion or an MRDC positive end of procedure outcome is
an RR authorization for the purposes of IRP.
EU laws which have been transposed into U.K. law through secondary legislation continue to be applicable as “retained
EU law.” However, new legislation such as the (EU) Clinical Trials Regulation will not be applicable in GB. Since a
significant proportion of the regulatory framework for pharmaceutical products in the United Kingdom covering the
quality, safety, and efficacy of pharmaceutical products, clinical trials, MAs, commercial sales, and distribution of
pharmaceutical products is derived from EU directives and regulations, Brexit may have a material impact upon the
regulatory regime with respect to the development, manufacture, importation, approval, and commercialization of our
product candidates in the United Kingdom. For example, the United Kingdom is no longer covered by the centralized
procedures for obtaining EU-wide MAs from the EMA, and a separate MA will be required to market our product
candidates in the United Kingdom. A new international recognition framework has been in place since January 1, 2024,
whereby the MHRA will have regard to decisions on the approval of MAs made by the EMA and certain other regulators
when determining an application for a new GB MA.
The medical device regulatory framework in Great Britain continues to be broadly based on the requirements of the (EU)
MDD as implemented into national law. On June 26, 2022, the MHRA published its response to a 10-week consultation on
the future regulation of medical devices in the U.K. Regulations implementing the new regime were originally scheduled
to come into force in July 2023, but the MHRA has confirmed that the core elements of the new framework are now
expected to be in place in 2025, while priority measures to enhance post-market surveillance will be put in place first in
2024. Medical devices bearing CE marks issued by EU notified bodies under the (EU) MDR or (EU) MDD are now subject
to transitional arrangements. Devices certified under the (EU) MDR may be placed on the market in GB under the CE
mark until June 30, 2030. However, devices certified under the (EU) MDD may be placed on the market until June 30,
2028. Following these transitional periods, it is anticipated that all medical devices will require a United Kingdom
Conformity Assessed ("UKCA") mark. Manufacturers may choose to use the UKCA mark on a voluntary basis prior to the
mandatory deadlines.
However, UKCA marking will not be recognized in the European Union. Following the transitional periods, compliance
with the United Kingdom regulations will be a prerequisite to be able to affix the UKCA mark to medical devices, without
which they cannot be sold or marketed in GB.
In addition, new regulations applicable in GB now require that all medical devices must be registered with the MHRA prior
to being placed on the market. Additionally, manufacturers based outside the United Kingdom will need to appoint a U.K.
Responsible Person to register devices with the MHRA.
Human Capital Resources
As of December 31, 2025, we had 1,120 full-time employees and 64 part-time employees. Of our 1,184 full- and part-time
employees, 18% have Ph.D. or M.D. degrees and 55% have graduate or post-graduate qualifications. 14% of our 1,184
employees are engaged in research and development activities and 73% are engaged in commercialization activities.
13% are engaged in global services activities including finance, legal, risk, people and culture, information technology.
Our human capital resources objectives include, as applicable, identifying, recruiting, retaining, incentivizing and
integrating our existing and additional employees. We support our employees by offering annual performance-based
bonus, equity-based incentive program, employee assistance programs, paid wellness days, hybrid work arrangements
and support for learning and development.
154
Legal Proceedings
See "Item 8. Financial Information - Consolidated Statements and Other Financial Information - Legal Proceedings."
Seasonality
We do not believe that seasonal influences have had a material effect on our business, financial condition, or results of
operations. The target disease indications for Illuccix and our other product candidates are not seasonal diseases.
Accordingly, once we have successfully obtained regulatory approvals to commercialize our other product candidates, if
ever, we do not anticipate that our business will be materially affected by seasonal influences in the future.
Australian Disclosure Requirements
Environmental regulation and compliance; Sustainability report
Telix seeks to be compliant with all applicable environmental laws and regulations relevant to its operations, including but
not limited to Australia, Belgium, Japan, Canada and the U.S. We monitor compliance on a regular basis to minimize the
risk of non‑compliance.
We conduct our activities at TMS in Brussels South, Belgium, in accordance with applicable environmental regulations,
including regular inspections by the Belgian Federal Agency for Nuclear Control ("FANC"). In 2022, TMS received
updated authorizations from FANC, aligned with the scope of Telix operations, and Telix is complying with its obligations
under these licenses and existing Belgian regulation. In December 2022, TMS was granted an environmental permit from
the Regional Authorities, valid until 7 October 2042, and in 2025 an updated operation authorization from FANC, valid
until 8 May 2040.
Telix also conducts its activities at, RLS Radiopharmacies, TMS Sacramento, Telix Targeting Technologies, and
IsoTherapeutics in the U.S., ARTMS in Canada, TMS North Melbourne in Australia, and TMS Yokohama in Japan in
accordance with applicable environmental regulations, including inspections by relevant authorities as required.
There were no known environmental breaches at Telix operations during the reporting period.
Information about Telix’s sustainability program, including for environmental matters, is detailed in the Company's
Sustainability report.
Beyond these matters, Telix is unaware of any environmental regulations matters applying to the Group's operating
activities that require disclosure.
155
1.Organizational Structure
The following table sets out for each of our subsidiaries, the state or jurisdiction of incorporation or organization,
percentage ownership and voting interest held by us (directly or indirectly through subsidiaries):
Name of Entity | State or Jurisdiction of Incorporation or Organization | Percentage Ownership and Voting Interest (%) | ||
Telix Pharmaceuticals Ltd | Australia | N/A | ||
Telix Pharmaceuticals (Innovations) Pty Ltd | Australia | 100 | ||
Telix Pharmaceuticals Holdings Pty Limited | Australia | 100 | ||
Telix Pharmaceuticals International Holdings Pty Ltd | Australia | 100 | ||
Telix Pharmaceuticals Australia Holdings Pty Ltd | Australia | 100 | ||
Telix Pharmaceuticals (ANZ) Pty Ltd | Australia | 100 | ||
Telix Pharmaceuticals (Corporate) Pty Ltd | Australia | 100 | ||
Telix Pharmaceuticals (Belgium) SRL | Belgium | 100 | ||
Telix Innovations SA | Belgium | 100 | ||
Telix Innovations Rph Participacoes Ltda | Brazil | 51 | ||
Telix Pharmaceuticals (Canada) Inc. | Canada | 100 | ||
Telix ARTMS Inc. | Canada | 100 | ||
Telix Pharmaceuticals (France) SAS | France | 100 | ||
Telix Pharmaceuticals (Germany) GmbH | Germany | 100 | ||
Rhine Pharma GmbH3 | Germany | 100 | ||
Therapeia GmbH & Co. KG | Germany | 100 | ||
Therapeia Verwaltungs-GmbH | Germany | 100 | ||
Telix Pharma Japan KK | Japan | 100 | ||
Telix Pharmaceuticals (NZ) Limited | New Zealand | 100 | ||
Telix Pharmaceuticals (Singapore) Pte Ltd | Singapore | 100 | ||
Telix Pharmaceuticals (Switzerland) GmbH | Switzerland | 100 | ||
Telix Pharmaceuticals (UK) Ltd | United Kingdom | 100 | ||
Lightpoint Surgical Ltd | United Kingdom | 100 | ||
Lightpoint Surgical Spain S.L. (Lightpoint Medical Espana SLU) | Spain | 100 | ||
Telix Pharmaceuticals (US) Inc. | Delaware | 100 | ||
Telix Optimal Tracers, LLC | Delaware | 100 | ||
Telix IsoTherapeutics Group, Inc. | Delaware | 100 | ||
Telix QSAM, Inc. | Delaware | 100 | ||
QSAM Therapeutics Inc. | Texas | 100 | ||
RLS (USA), Inc. | Delaware | 100 | ||
Las Vegas Radiopharmacy, Inc. | Delaware | 100 | ||
Telix Targeting Technologies, Inc. | Delaware | 100 | ||
ARTMS US, Inc. | Delaware | 100 |
156
Name of Entity | State or Jurisdiction of Incorporation or Organization | Percentage Ownership and Voting Interest (%) | ||
Telix Pharmaceuticals Holdings Pty Ltd | Australia | 100 | ||
Telix Pharmaceuticals International Holdings Pty Ltd | Australia | 100 | ||
Telix Pharmaceuticals Australia Holdings Pty Ltd | Australia | 100 | ||
Telix Pharmaceuticals (Innovations) Pty Ltd | Australia | 100 | ||
Telix Pharmaceuticals (ANZ) Pty Ltd | Australia | 100 | ||
Telix Pharmaceuticals (Corporate) Pty Ltd | Australia | 100 | ||
Telix Pharmaceuticals (NZ) Limited | New Zealand | 100 | ||
Telix Pharma Japan KK | Japan | 100 | ||
Telix Pharmaceuticals (Singapore) Pte Ltd | Singapore | 100 | ||
Telix Pharmaceuticals (US) Inc. | Delaware | 100 | ||
Telix Optimal Tracers LLC | Delaware | 100 | ||
Telix Pharmaceuticals (Canada) Inc. | Canada | 100 | ||
Telix Innovations SA | Belgium | 100 | ||
Telix Pharmaceuticals (Germany) GmbH | Germany | 100 | ||
Telix Pharmaceuticals (Switzerland) GmbH | Switzerland | 100 | ||
Telix Pharmaceuticals (Belgium) SRL | Belgium | 100 | ||
Lightpoint Surgical Ltd | United Kingdom | 100 | ||
Lightpoint Surgical Spain S.L. | Spain | 100 | ||
Rhine Pharma GmbH | Germany | 100 | ||
Therapeia GmbH & Co. KG | Germany | 100 | ||
Therapeia Verwaltungs-GmbH | Germany | 100 | ||
Telix Pharmaceuticals (France) SAS | France | 100 | ||
Telix Pharmaceuticals (UK) Ltd | United Kingdom | 100 | ||
Telix IsoTherapeutics Group Inc. | Delaware | 100 | ||
Telix ARTMS Inc. | Canada | 100 | ||
ARTMS US, Inc. | Delaware | 100 | ||
Telix QSAM, Inc. | Delaware | 100 | ||
QSAM Therapeutics Inc. | Texas | 100 | ||
Telix Innovations RPH Participações Ltda. | Brazil | 51 | ||
RLS (USA) Inc. | Delaware | 100 | ||
Las Vegas Radiopharmacy, Inc. | Delaware | 100 | ||
Telix Targeting Technologies, Inc. | Delaware | 100 |
2.Property, Plant and Equipment
Our principal headquarters are located in Melbourne, Australia where we lease office space and have a separate R&D
facility. The majority of our workforce is in the U.S., based out of our U.S. headquarters in Indianapolis, Indiana, or our
Dallas, Texas office, our R&D facilities in Angleton, Texas and Los Angeles and Sacramento, California, working remotely,
or in one of our network of radiopharmacies that we acquired in January 2025 as part of our acquisition of RLS. We also
maintain offices in Sydney and Brisbane, Australia, in Brussels, Herstal (near Liège), Belgium, in Geneva, Switzerland, and
in Kyoto, Japan, and have R&D facilities in South Brussels, Belgium, Yokohama, Japan, and Vancouver, Canada. We
believe that our current facilities are adequate to meet our ongoing needs and that, if we require additional space, we will
be able to obtain additional facilities on commercially reasonable terms.
For additional information on our property, plant and equipment, see Note 19 to our audited consolidated financial
statements included elsewhere in this Annual Report.
157
ITEM 4A. UNRESOLVED STAFF COMMENTS
None.
ITEM 5. OPERATING AND FINANCIAL REVIEW AND PROSPECTS
The following discussion and analysis are based upon and should be read together with our consolidated financial
statements and the accompanying notes and other financial information included elsewhere in this Annual Report. This
discussion includes both historical information and forward-looking information based upon current expectations that
involve risk, uncertainties and assumptions. Our actual results may differ materially from management’s expectations as
a result of various factors, including, but not limited to, those discussed in “Item 3. Key Information — D. Risk Factors”
and elsewhere in this Annual Report.
Our audited consolidated financial statements as of December 31, 2025 and 2024 and for the years ended December 31,
2025, 2024 and 2023 have been prepared in accordance with IFRS Accounting Standards.
A.Operating Results
Overview
We are a commercial-stage biopharmaceutical company focused on the development and commercialization of
therapeutic and diagnostic radiopharmaceuticals and associated medical technologies. Our prostate cancer portfolio
includes Illuccix and Gozellix, our commercially available 68Ga-labelled PSMA-PET imaging agents for prostate cancer.
The revenues generated from sales of Illuccix (and Gozellix in 2025), the costs associated with such sales and our
operating and other expenses resulted in a loss after tax of $7.1 million, and a profit after tax of $33.7 million and $4.2
million for the years ended December 31, 2025, 2024 and 2023, respectively.
We are leveraging our commercial revenues as a source of funding for the development of additional therapeutic and
diagnostic product candidates in our pipeline. These product candidates include TLX591-Tx, a therapeutic rADC, being
evaluated in a Phase 3 clinical trial for the treatment of patients with prostate cancer and two innovative imaging agents,
TLX250-Px for kidney (renal) cancer and TLX101-Px for brain cancer (glioma).
Our operations have been financed primarily through cash generated by our commercial operations and the issuance and
sale of ordinary shares. Our total comprehensive loss was $14.1 million for the year ended December 31, 2025. Our total
comprehensive income was $36.0 million and $1.7 million for the years ended December 31, 2024 and 2023,
respectively. We expect our expenses to increase as we continue our development of, and seek regulatory approvals for,
our product candidates. In addition, if and when we seek and obtain regulatory approval to commercialize additional
product candidates, we will also incur increased expenses in connection with commercialization and marketing of any
such product. Our total comprehensive income or loss may fluctuate significantly from period-to-period, depending on
the timing of our clinical trials and our expenditures on other research and development activities.
158
Results of Operations for the Fiscal Years Ended December 31, 2025, 2024 and 2023
The following table sets forth a summary of our Consolidated statement of comprehensive income or loss for the periods
presented.
Year ended December 31 | 2025 vs. 2024 | 2024 vs. 2023 | |||||
2025 | 2024 | 2023 | Change | Change | Change | Change | |
US$'000 | US$'000 | US$'000 | US$'000 | % | US$'000 | % | |
(Recast) | (Recast) | ||||||
(in thousands, except percentage and per share data) | |||||||
Continuing operations | |||||||
Revenue from contracts with customers | 803,794 | 516,551 | 332,978 | 287,243 | 56 | 183,573 | 55 |
Cost of sales | (377,356) | (180,388) | (123,445) | (196,968) | 109 | (56,943) | 46 |
Gross profit | 426,438 | 336,163 | 209,533 | 90,275 | 27 | 126,630 | 60 |
Research and development costs | (171,249) | (127,930) | (85,315) | (43,319) | 34 | (42,615) | 50 |
Selling and marketing expenses | (96,766) | (55,953) | (33,464) | (40,813) | 73 | (22,490) | 67 |
Manufacturing and distribution costs | (44,593) | (16,670) | (6,251) | (27,923) | 168 | (10,420) | 167 |
General and administration costs | (95,789) | (85,318) | (49,668) | (10,471) | 12 | (35,650) | 72 |
Other gains/(losses)(net) | 11,735 | 4,885 | (23,827) | 6,850 | 140 | 28,712 | * |
Operating profit | 29,776 | 55,177 | 11,008 | (25,401) | (46) | 44,169 | 401 |
Finance income | 5,826 | 7,180 | 671 | (1,354) | (19) | 6,509 | 969 |
Finance costs | (40,868) | (24,442) | (9,085) | (16,426) | 67 | (15,357) | 169 |
(Loss)/profit before income tax | (5,266) | 37,915 | 2,594 | (43,181) | (114) | 35,321 | 1,362 |
Income tax (expense)/benefit | (1,859) | (4,230) | 1,614 | 2,371 | (56) | (5,844) | * |
(Loss)/profit for the year | (7,125) | 33,685 | 4,208 | (40,810) | * | 29,477 | 700 |
(Loss)/profit for the year attributable to: | |||||||
Owners of Telix Pharmaceuticals Limited | (7,125) | 33,685 | 4,208 | (40,810) | * | 29,477 | 700 |
Other comprehensive (loss)/income: | |||||||
Items that will not be reclassified to profit or loss in subsequent periods: | |||||||
Changes in the fair value of investments at fair value through other comprehensive income | (1,242) | (3,287) | (612) | 2,045 | (62) | (2,675) | 437 |
Items to be reclassified to profit or loss in subsequent periods: | |||||||
Exchange differences on translation of foreign operations | (5,757) | 5,568 | (1,863) | (11,325) | * | 7,431 | * |
Total comprehensive (loss)/income for the year | (14,124) | 35,966 | 1,733 | (50,090) | * | 34,233 | 1,975 |
Total comprehensive (loss)/income for the year attributable to: | |||||||
Owners of Telix Pharmaceuticals Limited | (14,124) | 35,966 | 1,733 | (50,090) | * | 34,233 | 1,975 |
US$ Cents | US$ Cents | US$ Cents | |||||
Basic (loss)/earnings per share from continuing operations after income tax attributable to the ordinary equity holders of the Company | (2.11) | 10.17 | 1.32 | ||||
Diluted (loss)/earnings per share from continuing operations after income tax attributable to the ordinary equity holders of the Company | (2.11) | 9.76 | 1.30 | ||||
*Percentage not meaningful
159
Comparison of Years Ended December 31, 2025 and 2024
Revenue from Contracts with Customers
Revenue from contracts with customers was $803.8 million for the year ended December 31, 2025, an increase of
$287.2 million, or 56%, compared to $516.6 million for the year ended December 31, 2024. This increase was due to
growth in commercial sales from our Precision Medicine business as well as eleven months of revenue from RLS,
acquired on January 28, 2025.
Cost of Sales
Cost of sales increased by $197.0 million, or 109%, to $377.4 million for the year ended December 31, 2025 from $180.4
million for the year ended December 31, 2024. The increase was primarily driven by increased sales volumes and the
change in product mix following the acquisition of RLS.
Gross margin decreased in 2025 relative to 2024, decreasing to 53% for 2025 (compared to 65% in 2024). This decrease
was driven by the change in product mix following the acquisition of RLS and its sales of lower-margin SPECT imaging
products.
Research and Development Costs
R&D costs were $171.2 million for the year ended December 31, 2025, an increase of $43.3 million, or 34%, compared to
$127.9 million for the year ended December 31, 2024. This increase was primarily driven by:
•preparation for the re-submission of regulatory packages to the FDA for two new imaging agents in the U.S.
•production of inventory for commercial launches
•patient recruitment and clinical manufacturing for the Phase 3 ProstACT Global trial, and
•expanding our clinical trial activity and other R&D activity, as our current product candidates advance through
development and as we invest in future product candidates and programs.
Selling and Marketing Expenses
Selling and marketing expenses were $96.8 million for the year ended December 31, 2025, an increase of $40.8 million,
or 73%, compared to $56.0 million for the year ended December 31, 2024. This increase was primarily driven by
increased investment in promotional activities and sales force operations, deployed to drive higher sales volumes of
Illuccix, and to support the launches of Gozellix in the U.S. and Illuccix in Europe.
Manufacturing and Distribution Costs
Manufacturing and distribution costs were $44.6 million for the year ended December 31, 2025, an increase of $27.9
million, or 168%, compared to $16.7 million for the year ended December 31, 2024. This increase was primarily driven by
the impact of 11 months of RLS operations on the Group results, and higher costs associated with preparing the Brussels
South manufacturing facility for GMP commercial production.
General and Administration Costs
General and administration costs were $95.8 million for the year ended December 31, 2025, an increase of $10.5 million,
or 12%, compared to $85.3 million for the year ended December 31, 2024. This increase was driven by:
•the impact of 11 months of RLS operations on the Group results
•costs incurred to implement a Sarbanes-Oxley program, and
•professional fees in connection with the subpoena from the U.S. Securities and Exchange Commission (SEC)
Other Gains/(Losses) (Net)
Other gains (net) were $11.7 million for the year ended December 31, 2025, a change of $6.9 million, compared to other
gains (net) of $4.9 million for the year ended December 31, 2024. This resulted from a remeasurement of contingent
consideration on ARTMS and RLS based on the probability of achieving certain milestones, and unrealized foreign
currency gains.
Finance Income
Finance income was $5.8 million for the year ended December 31, 2025, a decrease of $1.4 million, or 19%, compared to
$7.2 million for the year ended December 31, 2024. Following the acquisition of RLS in January 2025 and associated
160
cash settlement of the transaction, there was a decrease in cash and cash equivalents placed into short term deposits in
the year ended December 31, 2025 compared to the prior year.
Finance Costs
Finance costs were $40.9 million for the year ended December 31, 2025, an increase of $16.4 million, or 67%, compared
to $24.4 million for the year ended December 31, 2024. This increase reflects the full year impact of interest paid and the
unwind of discount on Convertible Bonds issued in July 2024.
Income Tax Expense
Income tax expense was $1.9 million for the year ended December 31, 2025, a decrease of $2.3 million compared to an
expense of $4.2 million for the year ended December 31, 2024. This resulted from the recognition of $12.1 million in
deferred tax benefits attributable to temporary differences and unused tax losses. Current tax expense decreased from
$18.6 million in 2024 to $14.0 million in 2025 as a result of the decrease in taxable profits generated in the U.S. and
Belgium.
Comparison of Years Ended December 31, 2024 and 2023
Revenue from Contracts with Customers
Revenue from contracts with customers was $516.6 million for the year ended December 31, 2024, an increase of $183.6
million, or 55%, compared to $333.0 million for the year ended December 31, 2023. This increase was due to a 55%
increase in commercial sales volumes of Illuccix in the U.S. compared to 2023, supported by a combination of increased
market share and category growth for PSMA imaging, predominantly in the U.S.
Cost of Sales
Cost of sales increased by $57.0 million, or 46%, to $180.4 million for the year ended December 31, 2024 from $(123.4)
for the year ended December 31, 2023. The increase was primarily driven by higher dose administration fees to
distributors.
Gross margin improved in 2024 relative to 2023, increasing to 65% for 2024 (compared to 63% in 2023). This increase
was supported by a stable selling price for Illuccix® within each market segment.
Research and Development Costs
R&D costs were $127.9 million for the year ended December 31, 2024, an increase of $42.6 million, or 50%, compared to
$85.3 million for the year ended December 31, 2023. This increase was primarily driven by preparation for the
commercial launches, including scale-up of inventory, of three new imaging agents in the U.S. and patient recruitment
and clinical manufacturing for the Phase 3 ProstACT Global trial. We expect our R&D costs to continue to increase as we
expand our clinical trial activity and other R&D activity, as our current product candidates advance through development
and as we invest in future product candidates and programs.
Selling and Marketing Expenses
Selling and marketing expenses were $56.0 million for the year ended December 31, 2024, an increase of $22.5 million,
or 67%, compared to $33.5 million for the year ended December 31, 2023. This increase was primarily driven by
increased investment in promotional activities and sales force operations, deployed to drive higher sales volumes of
Illuccix, and preparation activities to support the launch of three new imaging products in the U.S.
Manufacturing and Distribution Costs
Manufacturing and distribution costs were $16.7 million for the year ended December 31, 2024, an increase of $10.4
million, or 167%, compared to $6.3 million for the year ended December 31, 2023. This increase was primarily driven by
increased personnel costs from the acquisition of ARTMS and IsoTherapeutics, and higher costs associated with
preparing the Brussels South manufacturing facility for GMP commercial production.
General and Administration Costs
General and administration costs were $85.3 million for the year ended December 31, 2024, an increase of $35.6 million,
or 72%, compared to $49.7 million for the year ended December 31, 2023. This increase was driven by investments in
corporate infrastructure to support the expansion of services assisting commercial operations in each region,
professional fees incurred in connection with our Nasdaq listing and transaction costs related to the acquisitions of
ARTMS Inc, IsoTherapeutics Group, and RLS (USA) Inc.
Other Gains/(Losses) (Net)
Other gains (net) were $4.9 million for the year ended December 31, 2024, a change of $28.7 million, compared to other
losses (net) of $23.8 million for the year ended December 31, 2023. This resulted from a lower remeasurement of
contingent consideration and unrealized foreign currency gains.
161
Finance Income
Finance income was $7.2 million for the year ended December 31, 2024, an increase of $6.5 million compared to $0.7
million for the year ended December 31, 2023. This increase reflects an increase in cash and cash equivalents placed
into short term deposits and higher interest rate yields obtained on deposits in the year ended December 31, 2024
compared to the prior year.
Finance Costs
Finance costs were $24.4 million for the year ended December 31, 2024, an increase of $15.3 million, or 169%, compared
to $9.1 million for the year ended December 31, 2023. This increase was due to a higher unwind of discount on
contingent consideration liability for 2024, reflecting the increased contingent consideration liabilities and finance costs
associated with the Convertible Bonds issued during the year.
Income Tax (Expense)/Benefit
Income tax expense was $4.2 million for the year ended December 31, 2024, a change of $5.8 million compared to a $1.6
million benefit for the year ended December 31, 2023. This resulted from the recognition of $14.3 million in deferred tax
benefits attributable to temporary differences and unused tax losses. Current tax expense increased from $10.9 million in
2023 to $18.6 million in 2024 as a result of the increase in taxable profits generated in the U.S. and Belgium.
Alternative Performance Measures
In reporting financial information, the Group presents alternative performance measures ("APMs") which are not defined
or specified under the requirements of IFRS. The Group believes that these APMs, which are not considered to be a
substitute for or superior to IFRS measures, provide stakeholders with additional useful information on the underlying
trends, performance and position of the Group and are consistent with how business performance is measured internally.
The alternative performance measures are not defined by IFRS and therefore may not be directly comparable with other
companies’ alternative performance measures. The key APMs that the Group uses are outlined below.
APM | Closest equivalent IFRS measure | Reconciling items to IFRS measure | Definition and purpose |
Income statement measures | |||
Adjusted earnings before interest, tax, depreciation and amortization (Adjusted EBITDA) | Profit/(loss) before income tax | Finance costs, income tax expense, depreciation and amortization, remeasurement of provisions, other income and expenses. | Used to help assess current operational performance excluding the impacts of non- operating expenditure, finance costs and finance income, depreciation and amortization and taxation expense. It is a measure that management uses internally to assess the performance of the Group’s segments and make decisions on the allocation of resources. |
Adjusted earnings before interest, tax, depreciation and amortization and research and development (Adjusted EBITDAR) | Profit/(loss) before income tax | Finance costs, income tax expense, depreciation and amortization, remeasurement of provisions, other income and expenses and costs associated with product development activities. | Used to assess the Group's performance excluding non-operating expenditure, finance costs and finance income, depreciation and amortization, taxation expense and product development activities. Included as a metric for LTVR targets in 2023, 2024 and 2025. |
Adjusted earnings before interest, tax, research and development (Adjusted EBITRD) | Profit/(loss) before income tax | Finance costs, income tax expense, remeasurement of provisions, other income and expenses and costs associated with product development activities. | Used to assess the Group's performance excluding non-operating expenditure, finance costs and finance income, taxation expense and product development activities. Included as a metric for LTVR targets in 2022. |
Balance sheet measures | |||
Net tangible asset per share | None | Net assets excluding intangible assets, deferred tax assets and right-of-use assets divided by the Group's weighted average number of ordinary shares on issue. | Disclosed in the Group's Appendix 4E as required by Rule 4.3A of the ASX listing rules. |
162
Outlined below is a reconciliation of the Group's APMs used to measure performance.
2025 | 2024 | 2023 | |||
$’000 | $’000 | $’000 | |||
Metric | Note | Operating segment | (Recast) | (Recast) | |
Operating profit | 29,776 | 55,177 | 11,008 | ||
Adjusting items: | |||||
Revenue from contracts with customers | 4 | Therapeutics | (9,273) | (6,226) | (3,496) |
Research and development costs | 5 | 171,249 | 127,930 | 85,315 | |
U.S. listing costs | 7 | - | 6,026 | - | |
Acquisition transaction costs | 7 | - | 5,750 | - | |
Depreciation and amortization | 9 | 21,506 | 4,851 | 4,485 | |
Other (gains)/losses net | 10 | (11,735) | (4,885) | 23,827 | |
Adjusted EBITDAR | 201,523 | 188,623 | 121,139 | ||
Product development revenue and costs | (161,976) | (121,704) | (81,819) | ||
Adjusted EBITDA | 39,547 | 66,919 | 39,320 |
Segments
Our three reportable segments are Precision Medicine, Therapeutics and Manufacturing Solutions.
We evaluate the performance of our segments based on Adjusted EBITDA, calculated as earnings before interest, tax,
depreciation and amortization, adjusted for the effects of the remeasurement of contingent consideration and other
gains and losses which may have an impact on the degree to which earnings reflect the results of core operations, such
as an impairment or impairment reversal where the impairment is the result of an isolated, non-recurring event.
Our management uses Adjusted EBITDA to assess the core operating performance of segments and to make decisions
about the allocation of resources. We also believe this measure provides useful information to users of our financial
statements by allowing for the assessment of underlying trends in our current operational performance by excluding the
impacts of non-cash sunk costs.
Precision Medicine
The Precision Medicine segment focuses on the commercial sales of Illuccix, Gozellix and other diagnostic products that
may obtain regulatory approvals. This segment includes royalties and sales of goods (which account for the majority of
our revenue from operations), as well as the sales and marketing expenses and costs of sales necessary to support
those revenues and research and development costs associated with development activities of our diagnostic pipeline.
163
The following table sets forth the results of operations for our Precision Medicine segment for the years ended
December 31, 2025, 2024 and 2023.
Year ended December 31, | 2025 vs. 2024 | 2024 vs. 2023 | |||||
2025 | 2024 | 2023 | Change | Change | Change | Change | |
US$'000 | US$'000 | US$'000 | US$'000 | % | US$'000 | % | |
(Recast) | (Recast) | ||||||
(in thousands, except percentage data) | |||||||
Revenue from contracts with customers | 621,930 | 508,508 | 329,205 | 113,421 | 22 | 179,304 | 35 |
Cost of sales | (222,750) | (178,263) | (123,445) | (44,487) | 25 | (54,818) | 31 |
Gross profit | 399,180 | 330,245 | 205,760 | 68,935 | 21 | 124,485 | 38 |
Research and development costs | (71,156) | (71,628) | (53,669) | 472 | (1) | (17,959) | 25 |
Selling and marketing expenses | (82,359) | (55,358) | (33,358) | (27,001) | 49 | (22,000) | 40 |
Manufacturing and distribution costs | (10,262) | (5,251) | (4,798) | (5,011) | 95 | (452) | 9 |
General and administration costs | (22,303) | (27,777) | (20,707) | 5,474 | (20) | (7,070) | 25 |
Other losses (net) | (3,552) | (5,976) | (24,497) | 2,424 | (41) | 18,521 | (310) |
Operating profit | 209,548 | 164,255 | 68,731 | 45,293 | 28 | 95,524 | 58 |
Other losses (net) | 3,552 | 5,976 | 24,497 | 26 | - | (18,521) | (310) |
Depreciation and amortization | 3,333 | 3,679 | 3,729 | (346) | (9) | (50) | (1) |
Adjusted EBITDA | 216,433 | 173,910 | 96,957 | 42,523 | 24 | 76,953 | 44 |
Comparison of Years Ended December 31, 2025 and 2024
For the year ended December 31, 2025, revenue from contracts with customers for our Precision Medicine segment
consisted of $621.6 million (2024: $508.4 million) in sales of goods and $0.3 million (2024: $0.1 million) in royalty
revenue. Sales of Illuccix and Gozellix in the U.S. were the main driver of the 22% increase in revenue from contracts with
customers for the Precision Medicine segment. Cost of Sales increased 25% year-over-year, driven by higher
manufacturing volumes, distribution costs, and radiopharmacy-related expenses. Gross profit declined slightly to 64% vs.
65% in the prior year, primarily reflecting a balance between commercial rebate levels, supply chain costs, and ongoing
operational efficiencies.
R&D expenses were $71.2 million in 2025, compared to $71.6 million in 2024. These similar numbers represent Telix's
commitment to furthering our investment in diagnostic programs globally.
Selling and marketing expenses were $82.4 million in 2025, compared to $55.4 million in 2024. The increase was
predominantly due to additional sales force, expanded commercial infrastructure and marketing expenditure incurred to
drive higher volumes and market share of our prostate imaging agents in the U.S. and Australia, Illuccix launch activities
in Europe and pre-launch activities for Zircaix and Pixclara.
Manufacturing and distribution costs were $10.3 million in 2025, compared to $5.3 million in 2024, reflecting an increase
in facility and staff costs to support supply chain and quality activities to deliver clinical and commercial products.
General and administration costs were $22.3 million in 2025, compared to $27.8 million in 2024, reflecting operating
leverage and stabilization in corporate activities and related overheads.
Adjusted EBITDA increased by $42.5 million, or 24% to $216.4 million for the fiscal year ended December 31, 2025, up
from $173.9 million in 2024, reflecting the growth in commercial revenues and stabilization in gross margins, partially
offset by higher operating expenditure.
Comparison of Years Ended December 31, 2024 and 2023
For the year ended December 31, 2024, revenue from contracts with customers for our Precision Medicine segment
consisted of $508.4 million (2023: $328.9 million) in sales of goods and $0.1 million (2023: $0.3 million) in royalty
164
revenue. Sales of Illuccix in the U.S. were the main driver of the 35% increase in revenue from contracts with customers
for the Precision Medicine segment.
Adjusted EBITDA increased by $77.0 million to $173.9 million for the year ended December 31, 2024, up from $97.0
million in 2023.
Therapeutics
The Therapeutics segment focuses on the development of our core therapeutic pipeline for commercialization. This
segment includes revenue received from license agreements prior to commercialization and research and development
services.
The following table sets forth the results of operations for our Therapeutics segment for the years ended December 31,
2025, 2024 and 2023.
Year ended December 31, | 2025 vs. 2024 | 2024 vs. 2023 | |||||
2025 | 2024 | 2023 | Change | Change | Change | Change | |
US$'000 | US$'000 | US$'000 | US$'000 | % | US$'000 | % | |
(Recast) | (Recast) | ||||||
(in thousands, except percentage data) | |||||||
Revenue from contracts with customers | 9,273 | 6,226 | 3,496 | 3,047 | 49 | 2,730 | 44 |
Cost of sales | (229) | - | - | (229) | * | - | * |
Gross profit | 9,044 | 6,226 | 3,496 | 2,818 | 45 | 2,730 | 44 |
Research and development costs | (98,039) | (55,877) | (31,258) | (42,162) | 75 | (24,619) | 44 |
Selling and marketing expenses | (1,434) | (88) | (106) | (1,346) | 1,525 | 18 | (20) |
Manufacturing and distribution costs | (4,238) | (22) | (69) | (4,216) | * | 46 | (208) |
General and administration costs | (3,606) | (137) | (185) | (3,469) | 2,533 | 49 | (35) |
Other gains (net) | 21 | - | - | 21 | * | - | 100 |
Operating loss | (98,252) | (49,898) | (28,122) | (48,354) | 97 | (21,776) | 44 |
Other gains (net) | (21) | - | - | (21) | * | - | 100 |
Depreciation and amortization | 278 | - | - | 278 | * | - | * |
Adjusted EBITDA | (97,995) | (49,898) | (28,122) | (48,097) | 96 | (21,776) | 44 |
*Percentage not meaningful
Comparison of Years Ended December 31, 2025 and 2024
For the year ended December 31, 2025, revenue from contracts with customers for our Therapeutics segment consisted
of $9.1 million (2024: $6.2 million) in R&D services revenue. The year-over-year change in revenue from contracts with
customers for our Therapeutics segment reflected higher investment in our R&D expenditure toward new therapeutic
product candidates in the year ended December 31, 2025, which resulted in higher R&D services revenue from the Grand
Pharma contract.
Research and development costs were $98.0 million in 2025, compared to $55.9 million in 2024, reflecting the increased
R&D investment towards patient recruitment and clinical manufacturing for the Phase 3 ProstACT Global trial, and an
increased investment into the development of our late-stage therapeutic assets.
Selling and marketing expenses were $1.4 million in 2025, compared to $0.1 million in 2024. The increase was
predominantly due to an increase in global medical affairs activities and investigator-initiated trial costs.
Manufacturing and distribution costs were $4.2 million in 2025 compared to nil in 2024, predominantly due to the
increase in CMC activities undertaken by our Manufacturing Solutions business to advance our therapeutics pipeline and
logistics costs in delivering clinical doses to patients.
165
General and administration costs were $3.6 million in 2025, compared to $0.1 million in 2024, reflecting a higher
allocation of corporate services provided to support the growth in activities, including an increase in business
development, finance, intellectual property and legal costs.
Adjusted EBITDA for the Therapeutics segment was a loss of $98.0 million in 2025, compared to $49.9 million in 2024,
reflecting the increase in activities as our late-stage therapeutics products progress through the clinical stages of
development.
Comparison of Years Ended December 31, 2024 and 2023
For the year ended December 31, 2024, revenue from contracts with customers for our Therapeutics segment consisted
of $6.2 million (2023: $3.5 million) in R&D services revenue. The year-over-year change in revenue from contracts with
customers for our Therapeutics segment reflected higher investment in our R&D expenditure toward new therapeutic
product candidates in the year ended December 31, 2024, which resulted in higher R&D services revenue.
Adjusted EBITDA for the Therapeutics segment was a loss of $49.9 million in 2024, compared to $28.1 million in 2023,
reflecting the increased R&D investment towards patient recruitment and clinical manufacturing for the Phase 3
ProstACT Global trial.
Manufacturing Solutions
The Manufacturing Solutions segment focuses on the operations of our vertically integrated supply chain and
manufacturing business and includes our production TMS facilities at Brussels South, North Melbourne, Sacramento and
Yokohama, ARTMS, IsoTherapeutics, and RLS Radiopharmacies. This segment comprises revenue generated from
distribution service fee revenue, as well as the sale of PET and SPECT products at RLS. Outside of RLS, revenue is
generated by the provision of contract manufacturing services to companies in the radiopharmaceutical industry, as well
as the operating expenses associated with our manufacturing solutions business.
The following table sets forth the results of operations for our Manufacturing Solutions segment for the fiscal years
ended December 31, 2025, 2024 and 2023.
Year ended December 31, | 2025 vs. 2024 | 2024 vs. 2023 | |||||
2025 | 2024 | 2023 | Change | Change | Change | Change | |
US$'000 | US$'000 | US$'000 | US$'000 | % | US$'000 | % | |
(Recast) | (Recast) | ||||||
(in thousands, except percentage data) | |||||||
Revenue from contracts with customers | 172,591 | 1,817 | 277 | 170,774 | 9,398 | 1,540 | 85 |
Inter-segment revenue | 72,514 | - | - | 72,514 | * | - | * |
Cost of sales | (222,853) | (2,125) | - | (220,728) | * | (2,125) | 100 |
Gross profit/(loss) | 22,252 | (308) | 277 | 22,560 | (7,325) | (585) | 190 |
Research and development costs | (5,813) | (425) | (388) | (5,388) | 1,267 | (37) | 9 |
Selling and marketing expenses | (12,973) | (507) | - | (12,467) | 2,461 | (507) | 100 |
Manufacturing and distribution costs | (30,093) | (11,397) | (1,384) | (18,696) | 164 | (10,013) | 88 |
General and administration costs | (12,021) | (3,977) | (2,475) | (8,044) | 202 | (1,502) | 38 |
Other gains (net) | 14,711 | 81 | - | 14,630 | * | 82 | 101 |
Operating loss | (23,937) | (16,533) | (3,970) | (7,404) | 45 | (12,563) | 76 |
Other gains (net) | (14,711) | (81) | - | (14,630) | * | (82) | 101 |
Depreciation and amortization | 16,933 | 856 | 157 | 16,077 | 1,878 | 699 | 82 |
Adjusted EBITDA | (21,715) | (15,758) | (3,813) | (5,957) | 38 | (11,945) | 76 |
*Percentage not meaningful
166
Comparison of Years Ended December 31, 2025 and 2024
For the year ended December 31, 2025, revenue from contracts with customers for our Manufacturing Solutions segment
consisted of $172.6 million (2024: $1.8 million) in services revenue, with the year-over-year increase related to
operations of the RLS business acquired in late January 2025.
Selling and marketing expenses were $13.0 million in 2025, compared to $0.5 million in 2024 reflecting the 11 months of
operations from the newly acquired RLS. These costs comprise predominantly amortization of acquired intangible assets
of $4.8 million, employment costs, sales commissions and travel costs related to sales and marketing personnel.
Manufacturing and distribution costs were $30.1 million and general and administration costs were $12.0 million for our
Manufacturing Solutions segment for the year ended December 31, 2025 (compared to $11.4 million and $4.0 million for
the year ended December 31, 2024, respectively). These increases were predominantly driven by increased personnel
and occupancy costs from the newly acquired RLS business, combined with increased activity to prepare the Brussels
South facility for GMP commercial production.
Other gains were $14.7 million in 2025, compared to $0.1 million in 2024, comprising the remeasurement of provisions
related to contingent consideration liabilities for ARTMS and RLS. The remeasurements were based on revised
probabilities applied to the achievement of certain commercial milestones.
For the year ended December 31, 2025, Adjusted EBITDA for the Manufacturing Solutions business was a loss of $21.7
million, compared to $15.8 million in the year ended December 31, 2024. The year-over-year change in Adjusted EBITDA
was driven by increased investment in our manufacturing, supply chain and logistics functions and the continued
buildout of our Brussels South, Yokohama and North Melbourne facilities.
Comparison of Years Ended December 31, 2024 and 2023
For the year ended December 31, 2024, revenue from contracts with customers for our Manufacturing Solutions segment
consisted of $1.8 million in services revenue compared to $0.3 million for the year ended December 31, 2023, with the
year-over-year increase related to operations of the IsoTherapeutics business acquired during the year.
Manufacturing and distribution costs were $11.4 million and general and administration costs were $4.0 million for the
year ended December 31, 2024 (compared to $1.4 million and $2.5 million for the year ended December 31, 2023,
respectively). These increases were predominantly driven by increased personnel and occupancy costs from the newly
acquired ARTMS and IsoTherapeutics businesses, combined with increased activity to prepare the Brussels South facility
for GMP commercial production.
For the year ended December 31, 2024, Adjusted EBITDA for the Manufacturing Solutions segment was a loss of $15.8
million, compared to $3.8 million in the year ended December 31, 2024. The year-over-year change in Adjusted EBITDA
was driven by increased investment in our manufacturing, supply chain and logistics functions and the continued
buildout of our Brussels South facility.
For more information on our segment reporting, see note 3 to our audited consolidated financial statements appearing
elsewhere in this Annual Report.
Recently Adopted Accounting Pronouncements
Refer to note 2.2 within our Material accounting policy information for a summary of recently adopted or yet to be
adopted accounting pronouncements.
Foreign Private Issuer Status
We report under the Exchange Act as a “foreign private issuer” under U.S. securities laws. In our capacity as a foreign
private issuer, we are exempt from certain SEC and Nasdaq requirements.
Consequently, we are not subject to all of the disclosure requirements applicable to U.S. domestic public companies. For
example, we are exempt from certain rules under the Exchange Act that impose certain disclosure obligations and
procedural requirements for proxy solicitations under Section 14 of the Exchange Act. In addition, at present, our
executive officers, the members of our board of directors and our principal shareholders are exempt from the reporting
and “short-swing” profit recovery provisions of Section 16 of the Exchange Act and the reporting rules under the
Exchange Act with respect to their purchases and sales of our securities. However, absent an exemption from the SEC,
following recent legislative changes that become effective on March 18, 2026, our officers and directors will be subject
to the insider reporting obligations under Section 16(a) of the Exchange Act, including the requirements to file Forms 3, 4
and 5, pursuant to the Holding Foreign Insiders Accountable Act enacted on December 18, 2025. Moreover, we are not
required to file periodic reports and financial statements with the SEC as frequently or as promptly as U.S. companies the
securities of which are registered under the Exchange Act. In addition, we are not required to comply with Regulation FD,
which restricts the selective disclosure of material information.
We may take advantage of these exemptions until such time as we are no longer a foreign private issuer. We will remain
a foreign private issuer until such time that 50% or more of our outstanding voting securities are held by U.S. residents
and any of the following three circumstances applies: (i) the majority of the members of either our board of directors or
167
our global management team are U.S. citizens or residents; (ii) more than 50% of our assets are located in the U.S.; or (iii)
our business is administered principally in the U.S.
We have taken advantage of certain reduced reporting and other requirements in this Annual Report. Accordingly, the
information contained herein may be different from the information you receive from other public companies.
B.Liquidity and Capital Resources
Prior to the fiscal year ended December 31, 2023, we incurred operating losses in each year since our founding. We
anticipate that as we expand through strategic acquisitions, increase our sales and marketing efforts and expand our
investment in R&D, we will need additional capital to fund our operations, which we may raise through a combination of
equity offerings, debt financings, strategic collaborations and other third-party funding arrangements. Our future liquidity
and capital resources will depend on product revenue from the successful continued commercialization of Illuccix and
Gozellix, revenue from any future products for which we obtain regulatory approval and the R&D costs and other
expenditure necessary to support these initiatives and future products. Our total comprehensive loss was $14.1 million,
for the year ended December 31, 2025. Our total comprehensive income was $36.0 million and $1.7 million for the years
ended December 31, 2024 and 2023, respectively. As of December 31, 2025, we had cash and cash equivalents of
$141.9 million and accumulated losses of $154.5 million. As of December 31, 2025, we held 52.8% of our cash in
Australian dollars, 39.7% in U.S. dollars, 5.5% in Euros, 0.3% in British pounds, 0.5% in Canadian dollars and 1.0% in Swiss
Francs.
Sources and Uses of Liquidity
Our operations have been financed primarily through cash generated by our commercial operations and the issuance and
sale of new ordinary shares and Convertible Bonds.
We intend to leverage our commercial revenues and the proceeds raised from the issuance of and sale of new ordinary
shares and Convertible Bonds as a source of funding for the development of additional therapeutic and diagnostic
product candidates in our pipeline, including conducting label-expanding trials across our portfolio of diagnostic imaging
agents and advancing clinical trials for our therapeutic product candidates. In the years ended December 31, 2025, 2024
and 2023, we received $774.2 million, $467.7 million and $311.8 million respectively, in receipts from customers, which
predominantly consisted of collections from sales from our Precision Medicine and Manufacturing Solutions businesses.
In the first quarter of 2022, we entered into two loan agreements whereby BNP Paribas agreed to lend us $6.8 million
and IMBC Group agreed to lend us $4.5 million. Each loan is denominated in Euros, in the amounts of €6.1 million and
€4.0 million, respectively, and have been translated to US$ based on the applicable exchange rate as of December 31,
2025. Each loan has a 10-year term and an interest rate of 1.85% per annum, payable monthly, and each is repayable in
96 monthly installments beginning at the end of a two-year grace period. As of December 31, 2025, the outstanding
balance of these facilities was $10.4 million (translated based on the applicable exchange rate as of December 31, 2025).
In connection with the loan agreement with BNP Paribas, we also entered a roll-over loan agreement whereby BNP
Paribas agreed to lend us an additional $2.3 million (€2.0 million, translated based on the applicable exchange rate as of
December 31, 2025). The loan has a two-year extendable term and a per annum interest rate calculated by adding the
eurozone interbank interest rate as of the determination date to a 1.5% margin, payable based on our choice of interest
period ranging from 1 month to 12 months for each advance (with a default interest period of three months if no
alternative is chosen), and it is repayable in full upon its expiration date. As of December 31, 2025, we have not drawn
down from this facility. We have used the borrowings from these loans in order to fund the renovation and
redevelopment of our Brussels South production facility.
On July 30, 2024 the Group completed the issue of $426,140,000 (A$650,000,000) in convertible bonds maturing in
2029. The bonds are convertible into fully paid ordinary shares in Telix Pharmaceuticals Limited. The initial conversion
price of the convertible bonds is A$24.78 per share, subject to anti-dilution adjustments set out in the final terms and
conditions of the convertible bonds. The net proceeds were $416,324,000, after transaction costs.
The convertible bonds will bear interest at a rate of 2.375 per cent per annum. Interest will be payable quarterly in
arrears on October 30, January 30, April 30 and July 30 in each year, beginning on October 30, 2024. The convertible
bonds will mature on or about July 30, 2029, unless redeemed, repurchased, or converted in accordance with their
terms. The holders have the option to redeem all or some of the convertible bonds on July 30, 2027 at their principal
amount together with accrued but unpaid interest accrued to that date. The convertible bonds are listed on the SGX.
Funding Requirements
We believe that our existing cash resources and cash that we expect to generate from sales from the Precision Medicine
and Manufacturing Solutions businesses will be sufficient to meet our projected operating expenses and capital
expenditure requirements for at least the next 12 months, as well as our anticipated longer-term operating cash
requirements and obligations. Our expectations regarding our short-term and long-term funding requirements are based
on assumptions that may prove to be wrong, and we may need additional capital resources to fund our operating plans
and capital expenditure requirements.
We expect our expenses to increase in connection with our ongoing activities, particularly as we continue the
commercialization of products for which we receive regulatory approval and continue clinical development of our
168
therapeutic product candidates. Accordingly, we will need to obtain substantial funding in connection with our continuing
operations. Until we can generate a sufficient amount of revenue from the sale of approved products, if ever, we expect
to finance our operating activities through cash generated from commercial sales, existing cash and cash equivalents
and financing activities, which may include equity offerings, debt financings, collaborations, strategic alliances and
licensing arrangements. To the extent that we raise capital through the sale of equity or convertible debt securities, the
ownership interest of our investors will be diluted, and the terms of these securities may include liquidation or other
preferences that adversely affect the rights of holders of ADSs. Debt financing, if available, may involve agreements that
include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making
capital expenditures or declaring dividends. If we raise funds through collaborations, strategic alliances or licensing
arrangements with third parties, we may have to relinquish valuable rights to our technologies, intellectual property,
future revenue streams or product candidates. If we are unable to raise funds through equity or debt financings when
needed, we may be required to delay, limit, reduce or terminate our product development or future commercialization
efforts or grant rights to develop and market product candidates that we would otherwise prefer to develop and market
ourselves.
Our present and future funding requirements will depend on many factors, including, among other things:
•The amount of revenue received from commercial sales of products for which we receive marketing approval;
•The initiation, progress, timing, costs and results of our clinical trials for our product candidates;
•The costs associated with in-licensing or acquiring assets to expand our pipeline, acquiring businesses or assets to
vertically integrate our supply chain and manufacturing and acquiring complementary business;
•The amount of milestones and royalties that we may be required to pay under existing acquisition and licensing
agreements;
•Costs associated with expanding our organization;
•The costs involved in filing patent applications and maintaining and enforcing patents or defending against claims of
infringement raised by third parties;
•The time and costs involved in obtaining regulatory approval for our product candidates and any delays we may
encounter as a result of evolving regulatory requirements or adverse results with respect to any of these product
candidates; and
•The costs of operating as a public listed company in both Australia and the U.S.
For more information as to the risks associated with our future funding needs, see “Item 3. Key Information — D. Risk
Factors.”
Cash Flows
The following table summarizes our cash flows for the periods presented:
1.Year ended December 31,
2025 | 2024 | 2023 | |
US$'000 | US$'000 | US$'000 | |
(Recast) | (Recast) | ||
(in thousands) | |||
Net cash (used in)/from operating activities | (17,293) | 27,490 | 14,273 |
Net cash used in investing activities | (285,921) | (86,687) | (16,839) |
Net cash (used in)/provided by financing activities | (3,719) | 416,791 | 7,091 |
Net (decrease)/increase in cash and cash equivalents | (306,933) | 357,594 | 4,525 |
Operating Activities
Net cash used in operating activities was $17.3 million during the year ended December 31, 2025. The primary source of
cash from operating activities was $774.2 million in receipts from customers, which predominantly consisted of
collections from sales from the Precision Medicine and Manufacturing Solutions businesses. The primary uses of cash in
operating activities were payments to suppliers and employees of $710.6 million. Other operating cash outflows included
$51.8 million in contingent consideration payments and $21.3 million in income tax payments.
169
Net cash from operating activities was $27.5 million during the year ended December 31, 2024. The primary source of
cash from operating activities was $467.7 million in receipts from customers, which predominantly consisted of
collections from sales of Illuccix. The primary uses of cash in operating activities were payments to suppliers and
employees of $418.3 million. Other operating cash outflows included $23.9 million in contingent consideration payments
and $2.0 million in income tax payments.
Net cash from operating activities was $14.3 million during the year ended December 31, 2023. The primary sources of
cash from operating activities were $311.8 million in receipts from customers, which predominantly consisted of
collections from sales of Illuccix. The primary uses of cash in operating activities were payments to suppliers and
employees of $280.4 million. Other operating cash outflows included $10.9 million in contingent consideration payments
to former ANMI shareholders and $6.9 million in income tax payments.
Investing Activities
Net cash used in investing activities was $285.9 million during the year ended December 31, 2025. The primary uses of
cash in investing activities were the acquisition of businesses. We invested $220.7 million in payments toward our
acquisitions of RLS and ImaginAb and $18.1 million in payments related to the acquisition of intellectual property
associated with FAP targeting candidates, and $25.7 million in property, plant and equipment purchases for the buildout
of our manufacturing facilities.
Net cash used in investing activities was $86.7 million during the year ended December 31, 2024. The primary uses of
cash in investing activities were $32.9 million in financial assets, which included a $31.0 million cash deposit into a cash
security account to establish a working capital facility. We also invested $20.7 million in payments toward our
acquisitions of IsoTherapeutics and ARTMS, $13.1 million in payments related to the acquisition of intellectual property
associated with QSAM, $8.4 million in payments toward the purchase of isotope raw material purchases and $9.1 million
in property, plant and equipment purchases for the buildout of our manufacturing facility in Belgium.
Net cash used in investing activities was $16.8 million during the year ended December 31, 2023. The primary uses of
cash in investing activities were $8.7 million in payments toward our acquisition of QSAM and strategic investment in
Mauna Kea and $6.3 million in property, plant and equipment purchases for the buildout of our manufacturing facility in
Belgium.
Financing Activities
For the year ended December 31, 2025, net cash used in financing activities totaled $3.7 million. Financing activity cash
flows included $1.8 million received from the issuance of new ordinary shares on the exercise of options previously
granted to employees and $5.2 million paid toward lease liabilities.
For the year ended December 31, 2024, net cash provided by financing activities totaled $416.8 million. Financing
activity cash flows included $0.7 million received from the issuance of new ordinary shares on the exercise of options
previously granted to employees, net proceeds of $427.9 million received from the issue of convertible bonds and $1.3
million paid toward lease liabilities.
For the year ended December 31, 2023, net cash provided by financing activities was $7.1 million. Financing activity cash
flows included $4.2 million received from the issuance of new ordinary shares on the exercise of options previously
granted to employees, proceeds of $4.2 million received from borrowings related to the loan facilities provided for the
construction of our manufacturing facility in Belgium and $1.3 million paid toward lease liabilities.
Off-Balance Sheet Arrangements
During the periods presented, we did not, and we do not currently, engage in off-balance sheet financing arrangements
as defined under SEC rules, such as relationships with other entities or financial partnerships, which are often referred to
as structured finance or special purpose entities, established for the purpose of facilitating financing transactions that
are not required to be reflected on our consolidated statement of financial position. In addition, we do not engage in
trading activities involving non-exchange traded contracts.
C.Research and Development, Patents and Licenses, etc.
For a discussion of our research and development activities, see “— A. Operating Results” and “Item 4. Information on the
Company — B. Business Overview.”
D.Trend Information
Our growth strategy and trends affecting our performance are detailed in “— A. Operating Results” and “Item 4.
Information on the Company — B. Business Overview.” For a discussion of uncertainties and certain factors that could
materially affect our business, see “Item 3. Key Information — D. Risk Factors.”
170
E.Critical Accounting Estimates
We believe that the accounting policies set out in Note 2 to the Consolidated Financial Statements involve a high degree
of judgment and complexity. Accordingly, these are the policies we believe are the most critical to aid in fully
understanding and evaluating our consolidated financial condition and results of our operations. See Note 2 to our
audited consolidated financial statements appearing elsewhere in this Annual Report for a description of our material
accounting policies and Note 2.28 to our audited consolidated financial statements appearing elsewhere in this Annual
Report for additional information on our key judgments and estimates.
Australian Disclosure Requirements
State of Affairs
There have been no significant changes in the state of affairs of the Group during the financial year ended December 31,
2025 other than as disclosed in this Annual Report.
Events subsequent to the end of the financial year
There were no subsequent events that required adjustment to or disclosure in the Australian Directors’ report (as filed
with the ASX) or the Financial statements of the Company for the year ended December 31, 2025.
171
ITEM 6. DIRECTORS, SENIOR MANAGEMENT AND EMPLOYEES
Start of the Remuneration report for Australian Disclosure Requirements
The Board of Directors ("Board") of Telix presents the Remuneration report, which details the Company's remuneration
policy and practice for Key Management Personnel ("KMP") for the financial year ended December 31, 2025. The Board
has determined that each KMP is an "executive officer" within the meaning of Rule 3b-7 of the Exchange Act.
KMP are defined as individuals with the authority and responsibility for planning, directing and controlling the activities of
the Group, either directly or indirectly. Telix’s 2025 Remuneration report covers both the Company's Non-Executive
Directors ("NEDs") and executive officers noted below during the year ended December 31, 2025 and up to the date of
this Annual Report.
This Remuneration report has been prepared in accordance with the Australian Corporations Act 2001 (Cth) for the
Group.
A. Directors and Senior Management
1.Key Management Personnel
The following table sets forth information regarding our directors and executive officers as of the date of this Annual
Report. The following table lists the names of our directors and executive officers. The business address for our directors
and executive officers is c/o 55 Flemington Road, North Melbourne, Victoria 3051, Australia.
Name | Age | Position | Term during 2025 |
Non-Executive Directors | |||
H Kevin McCann AO1 | N/A | Non-Executive Director and Chairman of the Board | January 1 - May 21 |
Tiffany Olson2 | N/A | Non-Executive Director and Chair of the Board | Full year |
Marie McDonald | 69 | Non-Executive Director | March 3 - December 31 |
Mark Nelson3 | 66 | Non-Executive Director | Full year |
Jann Skinner | 68 | Non-Executive Director | Full year |
Executive Officers/Executive KMP | |||
Christian Behrenbruch PhD MBA | 51 | Managing Director and Group Chief Executive Officer ("MD & CEO") | Full year |
Darren Smith FCPA MBA | 60 | Group Chief Financial Officer ("CFO") | Full year |
David Cade MD MBA | 57 | Group Chief Medical Officer ("CMO") | Full year |
Darren Patti PharmD | 54 | Group Chief Operating Officer (COO) | Full year |
1 Mr. Kevin McCann retired as Non-Executive Director and Chairman of the Board immediately following the Company's Annual General Meeting on May 21, 2025.
2 Ms. Olson was appointed Chair of the Board on May 21, 2025 following Mr. McCann’s retirement, and resigned from the Board effective February 3, 2026.
3 Mr. Nelson was appointed Interim Chair of the Board following Ms. Olson's resignation as Chair and Non-Executive Director, effective February 3, 2026.
The responsibilities of our Board are described in our Board Charter and Constitution, each of which is filed as an exhibit
to this Annual Report. Our executive officers are responsible for making and executing decisions that build value in
accordance with Board-approved delegated authorities.
The following is the biographical information of our current directors:
Directors
Mark Nelson has served as a Non-Executive Director since September 2017 and as Interim Chair since February 3, 2026.
Dr. Nelson has served as Chairman of the Caledonia Investments Group since January 2012, and as a Director of The
Caledonia Foundation since August 2002. He previously served as Chief Executive Officer and Co-Chief Investment
Officer of the Caledonia Investments Group from February 1992 to January 2012. He has also served as Director of
Kaldor Public Art Projects since October 2005, Governor of the Florey Neurosciences Institute since October 2007,
Director of the Mindgardens Neuroscience Network since February 2018 and Chairman of Art Exhibitions Australia since
2019. Dr. Nelson received his B.Sc. from the University of Melbourne, his M.Phil from the University of Cambridge and his
Ph.D. from the University of Melbourne. We believe Dr. Nelson’s qualifications and experience in capital, equity and
investment markets, including in the life sciences industry, qualify him to serve on our board of directors.
172
Christian Behrenbruch is one of our Co-Founders, has served as Group Chief Executive Officer since January 2017 and
joined our board of directors as Managing Director in January 2017. He has previously served as Chief Executive Officer
at Mirada Solutions from July 2001 to December 2002, President at CTI Molecular Imaging (now Siemens Healthcare)
from August 2003 to September 2006, Chief Executive Officer at Fibron Technologies, Inc. from June 2008 to December
2011 and Chief Executive Officer at ImaginAb, Inc from October 2007 to February 2015. He served as a Director at
Siemens Molecular Imaging Ltd from May 2005 to September 2006, Momentum Biosciences LLC from July 2007 to June
2009, Radius Health Ltd (now Adaptix Ltd) from May 2009 to February 2011, Factor Therapeutics Limited from October
2015 to May 2021 and Amplia Therapeutics Limited from May 2016 to February 2020, and he was the Chairman of Cell
Therapies Pty Ltd (a partnership with the Peter MacCallum Cancer Centre) from October 2012 to July 2014. Dr.
Behrenbruch holds a Doctor of Philosophy (PhD) in biomedical engineering from the University of Oxford, an executive
Master of Business Administration (MBA) jointly awarded from New York University, HEC Paris and the London School of
Economics (TRIUM Program) and a Juris Doctor from the University of Melbourne. Dr. Behrenbruch is a Fellow of
Engineers Australia in the management and biomedical colleges and a Graduate of the Australian Institute of Company
Directors. We believe Dr. Behrenbruch’s expertise and over 20 years of experience in healthcare entrepreneurship and
executive leadership qualify him to serve on our board of directors.
Marie McDonald has served as a Non-Executive Director since March 2025. Ms. McDonald serves as a Non-Executive
Director of Nanosonics Limited (since October 2016), Walter and Eliza Hall Institute of Medical Research (since October
2016) and Nufarm Limited (since March 2017), and served as a Non-Executive Director of CSL Limited between August
2013 and October 2025. Ms McDonald practised as a commercial lawyer, specializing in mergers and acquisitions and
corporate governance, at Ashurst Australia (previously Blake Dawson Waldron) from 1984 to 2014. She was a member of
the Australian Takeovers Panel from 2001 to 2010, and was also Chair of the Corporations Committee of the Business
Law Section of the Law Council of Australia (2012-2013), having previously been a deputy chair. Ms. McDonald received
her BSc (Hons) and LLB (Hons) from the University of Melbourne. We believe Ms. McDonald's significant experience in
financial markets, M&A, regulatory policy, remuneration, risk management and compliance qualify her to serve on our
board of directors.
Jann Skinner has served as a Non-Executive Director since June 2018. Ms. Skinner was a partner at
PricewaterhouseCoopers from 1987 to 2004. She has served as Chair of Create Foundation Limited since July 2024 and
Director since 2004. She also served as Non-Executive Director of QBE Insurance Group Limited from October 2014 to
May 2024 and Director of HSBC Bank Australia Limited from April 2017 to April 2023. Ms. Skinner is a Fellow of both
Chartered Accountants Australia & New Zealand and the Australian Institute of Company Directors. She received her
Bachelor of Commerce (BCom) from the University of New South Wales. We believe Ms. Skinner’s expertise in audit and
accounting and prior board experience qualify her to serve on our board of directors.
Group Executive Team
The following is the biographical information of our Group Executive Team ("GET"), which includes executive officers/
KMP. Not all members of the GET are executive officers/KMP. The individuals designated as executive officers/KMP are
indicated below with an *.
Christian Behrenbruch* is one of our Co-Founders, has served as Group Chief Executive Officer since January 2017
and joined our board of directors as Managing Director in January 2017. Biographical information for Dr. Behrenbruch is
included above under "Item 6. Directors, Senior Management and Employees — A. Directors and Senior Management —
Directors."
Darren Smith* has served as our Group Chief Financial Officer since August 2022. Previously, he was Global Chief
Financial Officer and Company Secretary at Sirtex Medical Ltd from June 2008 to March 2019, and Chief Financial Officer
of Waverley Council from April 2019 to August 2021. Mr. Smith has over 20 years of experience in executive finance and
general management experience across a broad range of industries, including life-sciences, for publicly listed, private,
international, and Australian government organizations. Mr. Smith holds a Master of Business Administration (MBA) from
the University of New South Wales in Australia and a Bachelor of Business (Accounting) from Western Sydney University.
He has been a Fellow Certified Practicing Accountant for 20 years.
David Cade* has served as our Group Chief Medical Officer since January 2024. Prior to transitioning to this role, he was
the Chief Executive Officer of our Asia Pacific operations from May 2021 to December 2023 and our Chief Business
Officer and Head of Investor Relations from October 2019 to April 2021. Previously, he served as Chief Medical Officer at
Sirtex Medical Limited from January 2007 to September 2017 and Chief Medical Officer at Cochlear Limited from October
2017 to September 2019. Dr. Cade received a Bachelor of Medicine and Bachelor of Surgery (MBBS) from Monash
Medical School and a Master of Business Administration (MBA) from Melbourne Business School and ESADE Business
and Law School Barcelona. He is also a Graduate of the Australian Institute of Company Directors.
Darren Patti* was appointed as our Group Chief Operating Officer in March 2024. Prior to transitioning to this role, he
was the Chief Operating Officer and General Manager of our Americas operations from March 2021 to March 2024.
Previously, he served as Vice President of Operations at Sofie Biosciences Inc. from November 2019 to March 2021, and,
preceding this role, he served in numerous other leadership capacities over his 15 year tenure at Sofie, including
managing high capacity PET manufacturing facilities and directing regional operations over multiple PET manufacturing
locations. Prior to joining Sofie, he worked in brachytherapy manufacturing with a small startup which was eventually
acquired by CR Bard. Dr. Patti has over 20 years of experience in radiopharmaceutical and device manufacturing with
expertise in network management and operations, including new radiopharmaceutical manufacturing, implementation

1 Ms. Moran-Adams and Ms. Crowe are advisors to the GET.
173
and compliance. Dr. Patti holds a Doctor of Pharmacy (Pharm.D.) from the University of Illinois at Chicago and a Bachelor
of Arts from Southern Illinois University at Carbondale. He is also an Authorized Nuclear Pharmacist and is a licensed
pharmacist in multiple states within the U.S.
Kevin Richardson serves as our Chief Executive Officer, Telix Precision Medicine. Mr. Richardson leads the development
of the Company’s diagnostics, global marketing and commercial operations in the U.S. and Canada. He has more than 25
years’ experience in the healthcare industry, including seven years focused in sales, marketing and business operations
in the radiopharmaceutical segment. Immediately prior to joining Telix, Mr. Richardson was the Chief Operating Officer of
UroShape Medical, a technology company which has developed and successfully commercialized a medical device for a
large, undertreated segment in the women’s health market. Prior to this, he spent seven years in the Americas division of
Sirtex Medical Ltd.
Mary Jessel serves as our Group Chief of Clinical Affairs. Dr. Jessel has over 15 years of pharmaceutical, biotechnology
and drug development experience and has proven expertise in building effective Medical Affairs infrastructure, launch
and product lifecycle strategies. She is passionate about innovation that improves unmet medical need and enhancing
the accessibility and comprehensibility of complex medical and scientific information. Dr. Jessel has led successful
product launches at Ionis Pharmaceuticals and Akcea, drug development at Alcamena Stem Cell Therapeutics, and drug
development research at the University of California, San Francisco in leukodystrophies. She holds a PhD in
Neuroscience from the University of Michigan and a MBA from Portland State University.
Richard Valeix serves as our Chief Executive Officer, Telix Therapeutics. Mr. Valeix leads the Company’s therapeutic
pipeline commercialization and business development. He has more than 20 years of pharmaceutical industry
experience, including radiopharmaceuticals, gained in senior executive leadership roles across a broad range of
therapeutic product areas. Previously, Richard worked at Advanced Accelerator Applications (AAA), a Novartis Company
(from January 2014 to April 2021) where he served in the roles of General Manager for France, Switzerland, Belgium,
Netherlands and Luxembourg, and Global Head of Marketing and Sales.
Raphaël Ortiz serves as our Chief Executive Officer, Telix International. Mr. Ortiz leads the “rest of world” commercial
operations for Europe, Middle East and Africa (EMEA), Asia Pacific (APAC) and Latin America regions. He joined Telix with
more than 20 years of pharmaceutical industry experience in a variety of roles, including in finance, business
development, marketing and sales, as well as general management in Europe, Latin America and Asia. Prior to joining
Telix, he worked at Advanced Accelerator Applications, a Novartis Company, and most recently in the role of Asia-Pacific
Cluster Head, setting up the radioligand therapy operations in the region.
Lena Moran-Adams serves as our Group General Counsel.1 Ms. Moran-Adams has over 25 years’ experience driving
proactive, results driven legal and compliance solutions worldwide, including more than 20 years’ experience in the
pharmaceutical industry in various country, regional and global leadership roles. Prior to joining Telix, Ms Moran-Adams
was the Head of Legal and Business Conduct, Intercontinental at Gilead Sciences and a Global Head of Legal at Novartis.
She is admitted to the bar and entitled to practice law in Australia, the United Kingdom and in New York.
Meredith Crowe serves as our Senior Vice President, People & Culture.1 Ms. Crowe has been with Telix since October
2021 and has more than 10 years experience driving people strategy in health and biotech, most recently in
organizational development at the Peter MacCallum Cancer Centre. Ms. Crowe holds a Masters of Design from Victoria
University and is an IECL Certified Executive Coach.
Company Secretary
Genevieve Ryan has served as our Company Secretary since December 2022. Ms. Ryan has over 20 years’ experience in
legal and governance roles, including with ASX-200 companies. Previously, she was General Counsel – Governance,
Corporate and Commercial at Orora Limited. Ms. Ryan was also previously Senior Legal Counsel and Alternate Company
Secretary at Australian Pharmaceutical Industries Limited (acquired by Wesfarmers Limited). Ms Ryan began her career
as a lawyer with law firm Ashurst. She holds a Bachelor of Laws (Honors) and Bachelor of Science (Honors). Ms Ryan is a
fellow of the Governance Institute of Australia and a graduate member of the Australian Institute of Company Directors.
1.Family relationships
There are no family relationships among any of our executive officers and our directors.
2.Arrangements for election of Directors and members of Management
There are no contracts or other arrangements pursuant to which directors have been or must be selected.
174
Executive KMP employment
All Executive KMP are employed on ongoing, permanent contracts and have notice period and cascading non-compete
and non-solicit clauses in their employment agreements. The maximum non-compete and non-solicit period is detailed
below with cascading time periods applicable to the MD & CEO and CFO:
Role | Notice period | Non-compete and non-solicit | Restricted area |
Dr. Behrenbruch (MD & CEO) | 6 months | 6 months | Australia/United Kingdom/European Union or U.S.; Victoria; Melbourne |
Mr. Smith (CFO) | 4 months | 6 months | Australia; Victoria; Melbourne |
Dr. Cade (CMO) | 4 months | 6 months | Australia; Melbourne |
Dr. Patti (COO) | 4 months | 6 months | U.S.; Australia, United Kingdom and European Union; states, provinces or territories within U.S. |
Employment may be terminated by either party on the provision of notice in the minimum period stated above. In the
event of termination for cause, Telix may terminate an Executive KMP’s employment immediately without notice.
B.Compensation
2.Executive KMP remuneration approach
2.1. Remuneration principles

Telix's remuneration principles are designed to:





Attract, motivate and retain talent in Telix's operating markets | Reward company performance and execution of Telix's strategy | Align the interests of employees with shareholders | Be simple and transparent |
175
2.2. Overview of remuneration elements
Total Fixed Remuneration ("TFR") | Short Term Variable Remuneration ("STVR") | Long Term Variable Remuneration ("LTVR") | |
Purpose | Attract and retain global talent capable of leading and delivering Telix’s strategy. | Reward achievement of annual corporate and business unit ("BU") objectives aligned to the delivery of Telix’s strategy. | Align remuneration with shareholder outcomes, rewarding achievement of long- term sustainable performance and shareholder value creation. |
Principles | Target 80-120% of the market median (see Peer Group below) considering experience and capability. | Reward achievement against annual corporate objectives | Rewards achievement of long term financial and product milestones. Use of notional exercise price delivers value with share price growth. |
Composition and delivery | Base salary and pension contributions paid over the year, and packaged benefits.1 | Annual performance incentive delivered as: •50% cash (approx. February the following year), and •50% deferred share rights ("STVR SRs") which vest approx. 12 months after the cash payment. | Performance Share Appreciation Rights ("PSARs") subject to achievement of 3- year performance and vesting conditions, as well as a service requirement. Stretch target set (maximum 150% of target). Vesting approx. 2 months after the end of the performance period with 2-year exercise period. |
Peer Group | Global listed companies in the health care sector with a focus on the biotechnology and pharmaceutical and health care supply industries. Companies are chosen based on market capitalization and revenue with Telix positioned near the median. | ||
TTR | Total Target Remuneration ("TTR") being the sum of TFR, STVR and LTVR. | ||
1 Australian Executive KMP can elect to cap their superannuation at the statutory superannuation maximum and receive the additional 11.5% (January 1 to June 30, 2025) and 12%
(July 1 to December 31, 2025) over the maximum as base salary. Refer to section x for full details in the 2025 statutory remuneration disclosures.
3.2025 Executive KMP remuneration
3.1. Executive summary - 2025 KMP remuneration
3.1.1. 2025 target remuneration
The following table summarizes the 2025 target remuneration packages for Executive KMP effective 1 January 2025, as
disclosed in the 2024 Remuneration report:
Dr. Behrenbruch (MD & CEO) | Mr. Smith (CFO) | Dr. Cade (CMO) | Dr. Patti (COO) | |
Base salary | A$799,092 | A$705,600 | A$539,000 | US$414,000 |
TFR | A$892,985 | A$788,508 | A$602,333 | US$434,700 |
TFR compa ratio1 | 0.85 | 0.99 | 0.76 | 0.82 |
STVR2 (% of base salary) | 110% | 65% | 65% | 65% |
STVR target value | A$879,001 | A$458,640 | A$350,350 | US$269,100 |
LTVR2, 3 (% of base salary) | 150% | 100% | 100% | 100% |
LTVR target value | A$1,198,634 | A$705,600 | A$539,000 | US$414,000 |
TTR | A$2,970,624 | A$1,952,748 | A$1,491,683 | US$1,117,800 |
TTR compa ratio1 | 0.38 | 0.52 | 0.40 | 0.45 |
1 Compa ratio is the TFR or TTR for Executive KMP as a proportion of the median (50th percentile) of the 2023 Mercer benchmarking data.
2 2025 variable remuneration as a percentage of base salary increased in line with Mercer's 2023 recommendation.
3 LTVR maximum opportunity is 150% of target (subject to achievement of the stretch financial performance condition).
176
3.1.2. 2025 remuneration delivery
3.1.3. 2025 target remuneration mix
The annualized remuneration elements at target for Executive KMP are as follows, noting that the TFR proportion is lower
for the COO role as it is performed in the U.S. where pensions are paid at a lower proportion than in Australia, where the
CFO and CMO are based:
3.2. 2025 Executive KMP remuneration framework
3.2.1. Total fixed remuneration ("TFR")
TFR incorporates base salary, pension contributions and packaged benefits. Due to regional variations for pensions and
packaged benefits, Telix uses base salary to determine STVR and LTVR targets to minimize regional impacts upon Total
Target Remuneration.
The Board’s target approach is to pay TFR within 80-120% of the market median, considering each Executive KMP's
experience and capability, and relativity to the market benchmark. The market median used to set 2025 remuneration
was based on a peer group of 40 companies (peer group) as part of the Mercer 2023 benchmarking review and
recommendation.
Executive TFR is set on appointment and reviewed annually in line with Telix’s performance review cycle and is subject to
Board approval.
Executive KMP receive TFR (base salary, pension contributions and packaged benefits) in equal cash installments paid
over the year. Australian Executive KMP can elect to cap their pension (superannuation) at the statutory superannuation
maximum and receive any additional amount over the maximum as base salary.
Fixed pay effective January 1, 2025 was disclosed in the 2024 Remuneration report and was aligned with Mercer's 2023
recommendation and the Board’s principles for Executive Remuneration. For 2025, TFR reflected 76-99% of the market
median (50th percentile) of the 2023 peer group.
177
3.2.2. Overview of variable remuneration
Variable remuneration includes STVR and LTVR. Both elements have an equity component which is subject to the
following terms:
Feature | Summary of terms common to STVR and LTVR Equity |
Treatment of vested equity after departure | The Board will automatically exercise vested unrestricted STVR Share Rights ("STVR SRs") and PSARs into American Depository Shares ("ADSs") or shares for departed Executive KMP who retain their STVR SRs or PSARs after exit. This will generally occur within 90 days of equity becoming unrestricted. |
Securities Dealing Policy | Testing and vesting is completed in accordance with the policy. All Executives must operate within local requirements and the Securities Dealing Policy. |
Equity Incentive Plan rules | STVR SRs and PSARs are granted in accordance with the Equity Incentive Plan rules (approved by shareholders at the 2024 AGM). |
Equity grants to the MD & CEO | Any equity grant to the MD & CEO is subject to shareholder approval. |
Dividend and voting rights | Unvested and vested but unexercised STVR SRs and PSARs have no dividend or voting rights and are held subject to Telix’s Securities Dealing Policy. |
Other activities | Treatment of STVR and LTVR equity is subject to Board discretion in the case of other events, such as (but not limited to) a change of control. |
Equity holding | Based on the Executive KMP’s location: •U.S. Executives receive equity based on USD values on the Nasdaq (that vests into ADSs) •Executives in all other locations receive equity converted to A$ and on the ASX that vests into ASX:TLX shares |
3.2.3. Short Term Variable Remuneration ("STVR")
STVR is designed to reward achievement of annual corporate objectives aligned to the delivery of Telix’s strategy.
Feature | Summary of 2025 STVR terms | ||||
Performance period | January 1 to December 31, 2025 | ||||
Delivery | Cash: 50% (paid in approx. February 2026), and Equity: 50% granted in STVR SRs after the 2025 full year results announcement and restricted for approximately 12 months from the cash payment (the restricted period).1 | ||||
Equity exercise price and vesting | STVR SRs have a nil exercise price and at vesting treatment is based on the Executive's location: •U.S STVR SRs are automatically vested into ADSs at the end of the restricted period •All other locations STVR SRs vest with a two-year exercise period. | ||||
Performance measures | Corporate Objectives, including financial and non-financial measures that maintain focus on underlying value creation within business operations, together with objectives relevant to each executive’s BU (except for MD & CEO). | ||||
Corporate objectives | Financial: Revenue Earnings Cost control Strategic | Dr. Behrenbruch 40% 15% 15% 30% | Mr. Smith 20% 7.5% 7.5% 15% | Dr. Cade 20% 7.5% 7.5% 15% | Dr. Patti 20% 7.5% 7.5% 15% |
BU objectives | - | 50% | 50% | 50% | |
Modifiers | The Board has the discretion to apply modifiers to either increase or decrease the STVR outcome based upon non-corporate objective obligations: contribution to good corporate governance, company values and market engagement, and driving a performance culture throughout the organization. | ||||
STVR SRs calculations | The number of STVR SRs Executive KMP receive is determined as 50% of their total STVR outcome, divided by the allocation value being the Volume Weighted Average Price (VWAP) for the 5 trading days after the release of the 2025 full year results. | ||||
General treatment to cash component on termination | Where employment is terminated for any reason prior to the payment date, the cash component is forfeited. | ||||
178
Feature | Summary of 2025 STVR terms | ||||
General treatment to Equity component on termination (the Board retains discretion to determine a different treatment) | Termination for cause during the performance or restricted period | Forfeited. | |||
Other circumstances such as death, disability, retirement, redundancy, mutually agreed separation or resignation | Where an Executive's employment is terminated •during the performance period: forfeited •during the restricted period: retained. | ||||
Reporting | See section 3.3.1 for 2025 overall outcomes. The 2026 AGM NOM will set out the MD & CEO allocation value and number of STVR SRs to be granted, subject to shareholder approval. | ||||
1 In certain circumstances the Board may determine that participants receive a cash equivalent value of the vested element after testing.
3.2.4. Long Term Variable Remuneration ("LTVR")
LTVR aligns Executive KMP remuneration with shareholder interests and outcomes, rewarding achievement of long-term,
sustainable performance and shareholder value creation.
Feature | Summary of 2025 LTVR terms | |
Performance period | Cumulative three-year period: January 1, 2025 to December 31, 2027 | |
Delivery | Performance Share Appreciation Rights (PSARs) provide the right to acquire Telix shares or ADSs equal in value to the gain above the notional 'exercise' price, subject to the satisfaction of performance conditions set by the Board, and terms and conditions that apply over the Performance Period. PSARs are granted based on Executive location: | |
U.S. in US$ on the Nasdaq; PSARs will vest and upon exercise convert into ADSs | Other locations: A$ on the ASX; PSARs will vest and upon exercise convert into shares | |
Notional exercise price | The volume weighted average price (VWAP) of Telix shares or ADSs over the 20 trading days following the 2024 full year results announcement (February 21 to March 20, 2025): | |
U.S.: US$19.99 on the Nasdaq | Other locations: A$28.67 on the ASX | |
Testing, vesting and exercise | PSAR performance is tested following audited financial results in approx. February 2028. The Board will determine the number of PSARs that vest (between 0 and 150% based on company achievement against the performance measures)1. Following vesting, Executive KMP have a two-year exercise period. The value delivered to Executives is the positive gain between the notional exercise price and the share/ADS price at the time of exercise. | |
Grant method | Granted at the maximum possible outcome (150% of target). Following testing, PSARs that do not meet the milestones are forfeited and are not subject to retesting. | |
Performance measures | Performance measure | Vesting outcome |
Financial (Adjusted EBITDAR) milestone2 | Less than US$1,058 million | - |
US$1,058 million | 25% | |
Greater than US$1,058 million and up to US$1,284 million | Straight line between 25% and 50% | |
Greater than US$1,284 million and up to US$1,450 million | Straight line between 50% and 100% | |
Product milestones | For details regarding the rationale and the strategic significance of the product milestones, refer to section 8.2.5 of the 2024 Remuneration report | |
Precision Medicine | Marketing authorization of an additional urology imaging asset in the U.S. | Achieved 17% Not achieved 0% |
Therapeutics | Interim results from 3 pivotal trials across 3 therapeutic compounds | Achieved 17% Not achieved 0% |
TMS | Inclusion of a Telix Manufacturing Solutions (TMS) site in a submission of a new commercial product | Achieved 8% Not achieved 0% |
TMS achieves a break-even profit and loss in any financial year within the period | Achieved 8% Not achieved 0% | |
LTVR calculations | The number of PSARs each Executive KMP receives is based on the concluded value, being the fair value price (independently determined using a Black Scholes valuation), adjusted for the probability of achievement of the non-market vesting conditions: | |
U.S.: on the Nasdaq: US$6.4716 concluded value and US$8.4413 fair value. | Other locations: on the ASX: A$10.7997 concluded value and A$14.0866 fair value | |
179
Feature | Summary of 2025 LTVR terms | |
Equity grant details | 2025 LTVR PSARs were granted to all Executive KMP on May 30, 2025 with the exception of the MD & CEO whose grant was made on June 13, 2025 following shareholder approval at the 2025 Annual General Meeting. | |
General treatment on termination (the Board retains discretion to determine a different treatment) | Departure reason | Treatment |
Termination for cause | Forfeited | |
Resignation | Generally a pro-rata retain is calculated on the portion of the first year of the measurement period served, remaining on-foot to the usual testing and vesting date.3 | |
Death, disability, retirement, redundancy and mutually agreed separation | ||
Reporting | The 2025 PSARs testing outcomes will be reported in the 2027 Remuneration report, with equity movements advised to the market via ASX disclosure and reported in the 2028 Remuneration report. | |
1 In certain circumstances the Board may determine that participants receive a cash equivalent value of the vested element after testing.
2 Refer to ITEM 5. OPERATING AND FINANCIAL REVIEW AND PROSPECTS for definition of these non-IFRS measures
3 See section 4.1.4 for changes to the pro rata calculation applicable for PSARs granted on or after January 1, 2026.
3.3. 2025 Executive KMP remuneration outcomes
3.3.1. Short Term Variable Remuneration (STVR) outcomes
Following the objectives, weightings and targets set for STVR performance at the commencement of the financial year,
the Corporate Objectives outcomes for 2025 are as follows:
Objective (Target %) | Details | Outcome | % STVR achieved |
Financial: Revenue augmentation and diversification (40%) | The 2025 Corporate Objectives were set prior to the acquisition of RLS, and as a result, RLS revenue is not included in either the target or actual revenue achievement disclosed in the STVR outcomes. Telix achieved revenue of US$631 million in 2025, excluding RLS. This outcome exceeded the 2024 result by 22% and was very close to the revenue target (in $) for 2025, but diversification targets were not achieved for 2025. | Below target | 20% |
Financial: 1)Earnings (15%); and 2)Cost control (15%) | 1)EBITDAR: Telix achieved an Earnings Before Interest, Tax, Depreciation, Amortization and Research and Development (EBITDAR) of US$200 million in 2025, below target. 2)Cost control: Telix invested US$171 million in Product Development, in line with plan. The circumstances that contributed to not meeting the revenue target also affected the ability to meet the EBITDAR target, which was not met. Research and development investment was aligned with our strategy to deliver our late stage pipeline and includes Zircaix inventory value (US$14 million) in preparation for launch activities. | Below target | 15% |
Non-financial: compliance (30%) | Substantial progress was made towards compliance requirements aligned to the Nasdaq listing. On target performance was achieved for a number of elements, but one was only partially achieved. | Below target | 25% |
Total | 60% | ||
Executive KMP STVR is determined by achievement against the Corporate Objectives, and the Business Unit Objectives
relevant to their position. The following details are provided for Other Executive KMP and performance against their
Business Unit objectives in 2025:
180
Executive | Business Unit Performance summary | Outcome | % achieved | |
Business Unit (of 100%) | Total (Corporate & BU) | |||
Mr. Smith (CFO) | Global services: progress made for commercial launch readiness of Zircaix and Pixclara and RLS services teams integrated into Global Services. Substantial progress on SOX compliance, strategic procurement capability in place, but uplift continues into 2026. | Below target | 65.00% | 62.50% |
Dr. Cade (CMO) | Equal measure between the business unit outcomes for both Precision Medicine and Therapeutics. Therapeutics: significant progress has been made across the therapeutic portfolio, however some milestones have experienced delays into 2026. Precision Medicine: achieved Illuccix registration in multiple EU countries, US approval of Gozellix, and filing completed in China. Pixclara and Zircaix received complete response letters (CRLs) from the FDA, which impacted market authorization timelines. | Below target | 67.50% | 63.75% |
Dr. Patti (COO) | Telix Manufacturing Solutions: significant progress towards reliable clinical dose delivery for late-stage therapies. High on-time delivery for Illuccix and Gozellix. Some delays seen in Therapeutic dose delivery and technology transfers. Significant progress on product quality and Environment, Health and Safety (EHS). | Below target | 70.00% | 65.00% |
In 2025 the Board assessed the STVR Corporate Objectives and Business Unit scorecards as well as the modifiers as
detailed in section 3.2.3. Following this assessment, the Board decided not to apply any discretion or modifier to any
Executive KMP STVR outcomes:
Executive | Target | Awarded | Performance against target | |||
Total | Cash | STVR SRs | Achieved | Forfeited | ||
Dr. Behrenbruch (MD & CEO) | A$879,001 | A$527,401 | A$263,700 | A$263,700 | 60.00% | 40.00% |
Mr. Smith (CFO) | A$458,640 | A$286,650 | A$143,325 | A$143,325 | 62.50% | 37.50% |
Dr. Cade (CMO) | A$350,350 | A$223,348 | A$111,674 | A$111,674 | 63.75% | 36.25% |
Dr. Patti (COO) | US$269,100 | US$174,915 | US$87,457 | US$87,457 | 65.00% | 35.00% |
3.3.2. Long Term Variable Remuneration (LTVR) outcomes
All Executive KMP held unvested 2023 PSARs as at December 31, 2025. The outcomes achieved are as follows:
Measure | Target | Performance Period Result (January 1, 2023 to December 31, 2025) | Weight at target | % vesting |
Adjusted EBITDAR (Earnings before interest, tax, depreciation, amortization and research and development costs) | Threshold: A$227 million Target: A$332 million Stretch: A$403 million | Over the three-year cumulative period, Telix achieved the stretch target with an adjusted EBITDAR of A$760 million, based on: 2023: A$181 million 2024: A$285 million 2025: A$294 million | Threshold 25% Target 50% Stretch 100% | 100% |
ProstACT Global Phase III interim read- out completed | Achieve milestone | Significant progress has been made toward this target; however, the milestone was not met by the end of the performance period, December 31, 2025. | 25% | Nil |
181
Measure | Target | Performance Period Result (January 1, 2023 to December 31, 2025) | Weight at target | % vesting |
Pre-pivotal trial (pre- IND) meeting completed with a major regulator for one of Telix’s rare disease therapy programs | Achieve milestone | The pre-pivotal trial (pre-IND) meeting was completed during the performance period for TLX-101, one of Telix's rare disease therapy programs. | 25% | 25% |
Overall vesting | 125% | |||
No discretion was applied by the Board to either alter the product milestones or adjust the vesting outcomes, the results
were endorsed as realized at the testing date (December 31, 2025).
The 2023 PSARs will vest in March 2026, following the audited results release and aligned with the Securities Dealing
Policy. The outcome and equity movements for each Executive KMP will be reported in the 2026 Remuneration report.
3.3.3. 2025 equity disclosures
Telix grants equity for retention and alignment of Executive KMP and shareholder interests. During 2025 Executives
received 2025 LTVR PSARs under the Equity Incentive plan (see sections 3.2.4 and 6.3).
The following equity vested to Executive KMP during 2025:
Equity type | Grant date | Executive KMP | Grant details | Vesting details | Exercise details | ||||
Type | # units | Date | Exercise price | Date | Resultant shares | Value exercised | |||
Talent equity1 | 05-Apr-22 | COO | Rights | 15,000 | 24-Apr-25 | A$0.00 | 05-May-25 | 45,000 | A$1,274,400 |
15-Jun-23 | 15,000 | ||||||||
26-Aug-24 | 15,000 | ||||||||
2022 PSARs | 05-Apr-22 | MD & CEO | PSARs | 139,672 | 06-Mar-25 | A$4.95 | n/a | n/a | - |
CMO | 78,189 | n/a | n/a | - | |||||
COO | 15,826 | n/a | n/a | - | |||||
2022 PSARs | 24-Oct-22 | CFO | PSARs | 77,912 | A$6.15 | n/a | n/a | - | |
2021 options | 21-Jul-21 | COO | Options | 100,708 | 28-Oct-22 | A$5.37 | 10-Mar-25 | 100,000 | A$2,254,000 |
1. Refer to section 2.4 of the 2024 Remuneration report regarding Talent equity granted to Dr. Patti prior to his appointment to an Executive KMP position.
The following equity was exercised by Executive KMP in 2025:
•As detailed above, Dr. Patti's talent equity was auto-exercised at vesting aligned to the terms of the offer. In
addition, Dr. Patti exercised 100,000 options at an exercise price of A$5.37.
•On November 5, 2025, Dr. Behrenbruch exercised 100,708 TLXO009 options with an exercise price of A$4.95.
These options vested on October 28, 2022 as detailed in section 12.2.2 and the ASX/SEC disclosure dated
November 7, 2025. Dr. Behrenbruch paid the exercise cost of $441,101 to receive the resultant 100,708 shares.
3.3.4. Equity held by Executive KMP during 2025
The following plans remain in the performance (restricted) period as at December 31, 2025 for current Executive KMP:
Equity type | Grant | Restricted period | Vesting date | Performance conditions | Exercise price |
Performance rights1 | 19-Jul-21 | 19-Jul-21 to 18-Jul-26 | 18-Jul-26 | Achievement of cumulative APAC revenue target within the restricted period | A$0.00 |
PSARs (2023 LTVR & LTI) | 02-May-23; 24-May-23 | 01-Jan-23 to 31-Dec-25 | 31-Dec-25 | Adjusted EBITDAR and product milestones. Refer section 3.3.2 | A$6.90 |
182
Equity type | Grant | Restricted period | Vesting date | Performance conditions | Exercise price |
Talent equity2 | 31-Oct-23 | 01-Nov-23 to 31-Dec-26 | 31-Dec-26 | Continued employment and high performance to drive Telix's success | A$0.00 |
01-Nov-23 to 31-Dec-27 | 31-Dec-27 | A$0.00 | |||
PSIRs | 08-Mar-24 | 01-Jan-24 to 31-Dec-26 | 31-Dec-26 | Adjusted EBITDAR and Revenue Refer section 5.4.2 of the 2024 Remuneration report | A$0.00 |
01-Jan-24 to 31-Dec-27 | 31-Dec-27 | Product milestone Refer section 5.4.2 of the 2024 Remuneration report | A$0.00 | ||
PSARs (2024 LTVR & LTI) | 21-Mar-24; 22-May-24 | 01-Jan-24 to 31-Dec-26 | Thursday, December 31, 2026 | Adjusted EBITDAR and product milestones. Refer section 5.3.1 of the 2024 Remuneration report | A$11.94 |
PSARs (2025 LTVR) | 30-May-25 | 01-Jan-25 to 31-Dec-27 | Friday, December 31, 2027 | Adjusted EBITDAR and product milestones. Refer section 3.2.4 | Non-US KMP on ASX: A$28.67 U.S. KMP on Nasdaq: US$19.99 |
1 Granted to Dr. Cade in his pre-KMP role as CEO, APAC.
2 Granted to Dr. Patti in his pre-KMP role as COO, Americas, refer to 2024 Remuneration report for full details.
3.3.5. 2025 realized remuneration
The following voluntary disclosure is made on a non-IFRS basis.
Please refer to statutory remuneration disclosures in section 3.5.
Executive KMP | TFR | STVR | Equity | Total |
Dr. Behrenbruch (MD & CEO) | A$892,985 | A$527,401 | A$430,959 | A$1,851,345 |
Mr. Smith (CFO) | A$788,508 | A$286,650 | A$380,541 | A$1,455,699 |
Dr. Cade (CMO) | A$602,333 | A$223,348 | A$362,464 | A$1,188,145 |
Dr. Patti (COO) | US$434,700 | US$174,915 | US$78,226 | US$687,841 |
Disclosures are based on the following assumptions:
•TFR: contractual base salary plus superannuation for Australian Executives and 401k pension contributions for
U.S. Executives
•STVR: total STVR (cash and equity components) for the 2025 performance year, as detailed in section 3.3.1. This
includes both the amount paid in cash in February 2026 and the amount paid in STVR SRs that are deferred to
approximately February 2027.
•Equity: the number of vested 2023 PSARs multiplied by the appreciation per PSAR - being A$11.20 (the closing
share price on December 31, 2025), less the exercise price of A$6.90. For Dr. Patti, his equity value in A$ has
been converted to US$ to determine the total based on the 12 month average exchange rate for the year.
3.4. 2025 Executive KMP remuneration alignment to shareholder wealth
In line with Telix’s remuneration principles and philosophy, performance measures are chosen to align Executive
KMP and shareholder interests and ensure variable remuneration is contingent on outcomes that grow and
protect long-term shareholder value.
Telix’s financial performance for 2021 to 2025 is summarized below, noting that Telix has retrospectively changed its
presentation currency from Australian Dollars to United States Dollars (USD or US$). Refer to
183
Type | Measure | 2025 | 2024 | 2023 | 2022 | 2021 |
Short-term measures | Revenue from contracts with customers ($'000) | 803,794 | 516,551 | 332,978 | 111,219 | 5,708 |
Net cash (used in)/ from operating activities ($'000) | (17,293) | 27,490 | 14,273 | (44,440) | (44,579) | |
Long-term measures (non- IFRS measures) | Adjusted EBITRD ($’000)1 | 180,017 | 183,772 | 116,654 | 2,627 | (26,766) |
Adjusted EBITDAR ($'000)2 | 201,523 | 188,623 | 121,139 | 6,365 | (22,879) | |
Other measures | (Loss)/profit before income tax ($'000) | (5,266) | 37,915 | 2,594 | (68,513) | (60,461) |
Basic (loss)/earnings per share (cents) | (2.1) | 10.2 | 1.3 | (23.3) | (21.4) | |
Net tangible assets per share ($) | (1.00) | 0.1600 | 0.0239 | 0.0224 | (0.1451) | |
Dividend per share ($) | - | - | - | - | - | |
Closing share price – ASX:TLX ($) | A$11.20 | A$24.61 | A$10.08 | A$7.27 | A$7.75 | |
Increase/(decrease): ASX share price (%) | (54) | 144 | 39 | (6) | 105 | |
Closing ADR/ADS price – Nasdaq:TLX (US$) | US$7.49 | US$15.40 | n/a | n/a | n/a | |
Increase/(decrease): Nasdaq ADS price (%) | (51) | n/a | n/a | n/a | n/a | |
Market capitalization (US$'000) | 2,537,440 | 5,154,757 | 2,169,352 | 1,558,123 | 1,603,079 |
1. Adjusted EBITRD (Earnings Before Interest, Taxes and R&D expense) on a 3-year cumulative basis is the 2022 LTVR financial metric.
2. Adjusted EBITDAR (Earnings Before Interest, Taxes, Depreciation and Amortization and R&D expense) is the LTVR financial metric from 2023 onwards.


1 Remuneration includes movement in annual leave and long service leave provisions during the year.
2 Mr. Smith participated in the 2025 Australian ESPP and received a discount of $2,352 on shares acquired related to salary sacrifice in the period April to May 2025, allocated in September 2025. This discount is reflected in the assessed fair value of the ESPP rights
and disclosed in the Share-based payment column above.
3 Dr. Patti was appointed as Group Chief Operating Officer on March 11, 2024. His remuneration in 2024 is reported to include all amounts associated with his role as KMP from March 11, 2024.
4 Mr. Valeix moved to the non-KMP role of CEO, Telix Therapeutics, and ceased as KMP on August 18, 2024. His 2024 remuneration is reported to include all amounts associated with his role as KMP up to August 18, 2024. Mr. Valeix was paid in CHF, reported in USD
using the respective monthly exchange rates.
184
3.5. 2025 Executive KMP statutory remuneration
The below table shows details of the remuneration expenses recognized for Executive KMP for 2025 and 2024 prepared in accordance with IFRS and Australian Accounting
Standards.
All numbers have been recast for 2024 and provided in 2025 in US$ using the average exchange rate over the 2025 year.
Fixed remuneration1 | Variable remuneration | Termination benefit | Total | Variable remuneration | ||||||
Salary | Superannuation /pension | Leave accruals | STVR | Share-based payment | ||||||
Name | Year | US$ | US$ | US$ | US$ | US$ | US$ | US$ | US$ | % |
Executive KMP | ||||||||||
Dr. Behrenbruch | 2025 | 556,718 | 19,333 | (53,987) | 169,937 | 569,657 | - | 1,261,658 | 739,594 | 58.62 |
2024 | 400,134 | 18,961 | 3,325 | 155,983 | 350,187 | - | 928,590 | 506,171 | 54.51 | |
Mr. Smith2 | 2025 | 488,989 | 19,333 | 35,623 | 92,363 | 381,828 | - | 1,018,135 | 474,191 | 46.57 |
2024 | 350,817 | 18,896 | 26,345 | 66,748 | 341,589 | - | 804,395 | 408,337 | 50.76 | |
Dr. Patti3 | 2025 | 381,877 | 9,569 | 15,508 | 87,458 | 354,833 | - | 849,244 | 442,291 | 52.08 |
2024 | 288,000 | 20,270 | - | 64,920 | 236,072 | - | 609,262 | 300,992 | 49.40 | |
Dr. Cade | 2025 | 368,899 | 19,333 | 13,898 | 71,966 | 71,103 | - | 545,199 | 143,069 | 26.24 |
2024 | 340,573 | 18,961 | 21,643 | 64,894 | 259,382 | - | 705,452 | 324,275 | 45.97 | |
Former Executive KMP | ||||||||||
Mr. Valeix4 | 2025 | - | - | - | - | - | - | - | - | - |
2024 | 255,255 | 27,885 | 157 | 52,314 | 342,669 | - | 678,280 | 394,983 | 58.23 | |
Total | 2025 | 1,796,483 | 67,568 | 11,042 | 421,724 | 1,377,421 | - | 3,674,236 | 1,799,145 | 48.97 |
2024 | 1,634,779 | 104,973 | 51,470 | 404,859 | 1,529,899 | - | 3,725,979 | 1,934,758 | 51.93 | |
185
4. 2026 Executive KMP remuneration
Telix's headcount, revenue and market capitalization in early 2025 were significantly higher than the data used by the
prior Mercer benchmarking prepared in 2023.
Recognizing Telix's growth and development since the 2023 data was provided, in 2025 the Board engaged Mercer to
provide current remuneration benchmarking data for Executive KMP. A global peer group of 40 companies was
established with Telix positioned near the median for metrics such as market capitalization and revenue, based on 6
month average data to June 30, 2025. Given the changes in Telix’s share price during the year, updated data as to Telix’s
positioning in the peer groups, as at the end of October 2025, was also obtained.
The June 30 data indicated that current executive KMP remuneration was significantly below the benchmark median.
Executive KMP remuneration remained below the market midpoint, with both TFR and TTR midpoints increasing from the
2023 to 2025 benchmarking data. This movement resulted in further decreased compa ratios for remuneration going into
2026 (based on June 2025 data) compared with those in 2025 (based on the 2023 data).
The October 2025 data also confirmed that remuneration remained below Telix’s then position in the global peer group.
4.1. 2026 Executive KMP remuneration framework
4.1.1. Approach in 2026
Having regard to the shareholder experience in 2025, the Board decided not to make any material changes to KMP
remuneration for 2026, other than as mentioned below.
The Board decided to apply the average Telix employee market adjustment of 3.5% to the base salaries of Executive
KMP (including the CEO), with the exception of Dr. Patti who received a 15% uplift. This is in recognition of the significant
increase in position scope of his Group COO role to include radiopharmacy site and distribution network management
(including responsibility for approximately 500 additional employees) as a result of the RLS acquisition.
See section 4.2 for 2026 remuneration at target for Telix's Executive KMP.
4.1.2. 2026 remuneration delivery
No changes will be made in the delivery of remuneration to Executive KMP in 2026. Despite the positioning against the
benchmarking data outlined above, the Board determined that no changes would be made to STVR and LTVR
opportunity levels for 2026.
2026
2027
2028
2029
2030
2031


TFR
Equal monthly or 2
weekly installments

Performance
period
STVR
Deferral period


Cash (50%)
Equity (50%)



LTVR
Performance period
Exercise period
Performance testing
Payment / vesting


186
4.1.3. Short Term Variable Remuneration (STVR)
To promote alignment across the organization, in 2026 we are returning to having the same set of measures for STVR for
all GET members, consisting of corporate financial objectives (40%) and shared business unit objectives (60%). This is a
move away from the 2025 approach of individual business unit objectives assigned to each Executive KMP (excluding
the MD & CEO).
As a result, from January 1, 2026, STVR outcomes for all Executive KMP (including the MD & CEO) will be measured
based on achievement of a single Corporate objective scorecard comprising:
•Global revenue augmentation and diversification (20%),
•EBITDAR earnings target (10%),
•Product development (10%), and
•Business Unit priorities of 60%, split equally between Precision Medicine, Therapeutics, TMS and the
International Business
The Business Unit priorities are:
•Precision Medicine: a combination of revenue diversification and Px program milestones
•Therapeutics: patient enrollment into clinical trials, achieving trial approvals from regulators and data readouts
•TMS: effective delivery of Telix doses and commercial products, build R&D capability and increase capabilities
across global sites
•International Business: ex-U.S./Canada revenue and market share targets and trial registration, lodgment of
approvals and commercial availability of products within EMEA
The Board will continue to consider modifiers based on non-corporate objective obligations of Executive KMP as detailed
in section 3.2.3.
4.1.4. Long Term Variable Remuneration
The 2026 LTVR will replicate the 2025 approach as detailed in section 3.2.4, with elements included below where the
terms differ:
Feature | Summary of 2026 LTVR terms | |
Performance period | January 1, 2026 to December 31, 2028 | |
Notional exercise price | The volume weighted average price (VWAP) of Telix shares or ADSs over the 20 trading days following the 2025 full year results announcement (February 23 to March 20, 2026). | |
Testing, vesting and exercise | PSAR performance will be tested following the audited financial results in March 2029. The Board will determine the number of PSARs that vest (between 0 and 150% based on the performance measures)1. Following vesting, Executive KMP have a two-year exercise period for the vested PSARs. The value received to Executives is the positive gain between the notional exercise price and the share/ADS price at the time of exercise. | |
Grant method | Granted at the maximum possible outcome (150%), where the stretch financial target is achieved alongside all product milestones. Following testing, PSARs are forfeited that don’t meet the milestones and are not subject to retesting. | |
Performance measure | Performance measure | Vesting outcome |
Financial (Adjusted EBITDAR) milestones | Less than US$1,365 million | - |
US$1,365 million | 25% | |
Greater than US$1,365 million and up to US$1,837 million | Straight line between 25 and 50% | |
Greater than $1,837 million and up to US$2,042 million | Straight line between 50 and 100% | |
Product milestones | ||
187
Feature | Summary of 2026 LTVR terms | |
Precision Medicine (Px) | Gain FDA approval for a sNDA or a NDA of a new asset or indication expansion of a PSMA targeting imaging agent. | If achieved 12% If not achieved 0% |
Submit for a non-prostate indication expansion (metatastic) in the Precision Medicine business unit | If achieved 4% If not achieved 0% | |
Therapeutics (Tx) | Submit a marketing authorization for a TLX therapeutic product in a commercially relevant jurisdiction | If achieved 15% If not achieved 0% |
Run an EAP/NPP with government financial support, of one Tx asset in a relevant jurisdiction | If achieved 2% If not achieved 0% | |
Telix Manufacturing Solutions (TMS) | Have at least five RLS sites manufacturing a commercial Telix Px imaging agent. | If achieved 6% If not achieved 0% |
Delivery of at least four Tx drug product production lines within the US market capable of Phase 3/commercial production | If achieved 6% If not achieved 0% | |
Delivery of at least one commercial ready therapeutic isotope supply chain | If achieved 5% If not achieved 0% | |
Equity grant details | 2026 LTVR PSARs will be granted at stretch target (150% outcome) to all Executive KMP, with the MD & CEO grant made after shareholder approval at the 2026 Annual General Meeting. | |
Treatment on termination (the Board retains discretion to determine a different treatment) | Departure reason | Treatment |
Termination for cause | Forfeited | |
Resignation | Generally the number of PSARs retained is calculated on a pro rata basis with the remaining PSARs staying on foot to the usual testing and vesting date. The pro rata calculation and % retained is as follows: •Up to 12 months: 0% •12 months and less than 24 months: 25% •24 months and less than 36 months: 50% •Full three-year period: 100% retained | |
Death, disability, retirement, redundancy and mutually agreed separation | A pro-rata portion of the unvested PSARs based on the number of days between the beginning of the Performance Period and the exit date will remain on foot to the usual testing and vesting date. | |
Reporting | The 2026 PSARs testing outcomes will be reported in the 2028 Remuneration report, with equity movements advised to the market via ASX disclosure and reported in the 2029 Remuneration report. | |
1 In certain circumstances the Board may determine that participants receive a cash equivalent value of the vested element after testing.
188
Additional information on 2026 LTVR performance measures
Measure | Rationale and strategic significance | Calculation | Measure type |
Adjusted EBITDAR | Demonstrates Telix’s underlying performance before non-operating expenditure, finance costs, depreciation and amortization, taxation expense and research and development activities. | Refer to Alternative performance measures | Financial |
Precision Medicine (Px) | Expansion of the Telix PSMA franchise and non-prostate indication expansion will increase Telix's opportunity for continued sales growth and strengthened market position. | Either achieved or not achieved milestone (hit/ miss). | Strategic delivery |
Therapeutics (Tx) | Regulator engagement with Therapeutic assets such as an EAP/NPP and marketing authorization submission shows key progress of Telix's Therapeutic assets. | ||
TMS | Supply and qualification of both Precision Medicine and Therapeutic production capabilities within RLS demonstrates successful expansion, and the maturation of TMS sites aligning with projected commercial needs. | ||
Targets | The Board sets targets at the outset of each performance period. Targets are set to be sufficiently challenging for Executive KMP and deliver appropriate returns for shareholders. The product milestones reflect Telix’s strategy and focus on the Px, Tx and TMS business units. Including these measures for Executive KMP ensures a cohesive approach across the Executive team, towards sustainable company and shareholder long term value. | ||
4.2. 2026 Executive KMP remuneration at target
Executive KMP remuneration from January 1, 2026 is outlined below. No changes were made to STVR and LTVR
opportunity levels.
Dr. Behrenbruch (MD & CEO) | Mr. Smith (CFO) | Dr. Cade (CMO) | Dr. Patti (COO) | |
Base salary | A$827,060 (3.5% uplift) | A$730,296 (3.5% uplift) | A$557,865 (3.5% uplift) | US$476,100 (15% uplift) |
TFR | A$926,307 | A$817,932 | A$624,809 | US$499,905 |
TFR compa ratio1 | 0.68 | 0.85 | 0.70 | 0.77 |
STVR2 (% of base salary) | 110% | 65% | 65% | 65% |
STVR target value | A$909,766 | A$474,692 | A$362,612 | US$309,465 |
LTVR2, 3 (% of base salary) | 150% | 100% | 100% | 100% |
LTVR target value | A$1,240,590 | A$730,296 | A$557,865 | US$476,100 |
TTR | A$3,076,664 | A$2,022,920 | A$1,545,286 | US$1,285,470 |
TTR compa ratio1 | 0.56 | 0.52 | 0.42 | 0.48 |
1 Compa ratio is the TFR for an Executive KMP as a proportion of the median (50th percentile) of the recent 2025 Mercer benchmarking data.
2 Variable remuneration as a percentage of base salary increased in line with Mercer's recommendation (STVR and LTVR).
3 LTVR maximum opportunity is 150% of target.
5. 2025 NED remuneration
5.1. Remuneration principles
To attract and retain suitably qualified individuals, NED fees are set to reflect the obligations, responsibilities and
demands of Telix’s NEDs. They are reviewed periodically by the Board, considering market benchmark data and the
financial position of the Group.
As detailed in the 2024 Remuneration report, increases were made to NED remuneration effective January 1, 2025 to
align with the 80 to 120% of the market median approach, utilizing the data provided by Mercer in 2023 (not a
recommendation). Mercer’s 2023 benchmarking considered 19 ASX listed companies, with Telix positioned towards the
median based on the average six-month market capitalization at August 31, 2023.
NEDs receive fees as Directors of Telix and for membership or chairing of some Board Committees. NEDs do not receive
any performance-based remuneration. The Chair is not compensated for Committee memberships but is compensated
for Committee Chair roles, where applicable.
189
The most recent NED Share Appreciation Rights ("NED SARs") grant made to Ms. Olson after shareholder approval at the
2022 Annual General Meeting vested on May 19, 2025.
As disclosed in the 2024 Remuneration report, during 2025 the Board introduced:
•A minimum shareholding policy for NEDs to hold equity equivalent to their annual base NED fee within 3 years of
appointment or the introduction of the policy in 2025, and
•NED Rights plans for directors to salary sacrifice up to 40% of their base Board fees into Telix equity
There is no retirement benefit scheme (other than statutory superannuation contributions as required for Australian-
based NEDs).
5.2. 2025 NED remuneration
The NED aggregate fee limit of A$1,800,000 was approved by shareholders at the 2025 Annual General Meeting. Total
NED remuneration paid during 2025 was A$744,699, within the fee limit (41.4% of the total).
NED remuneration (inclusive of superannuation or other relevant statutory requirements, as applicable) are as follows:
Board and Committee Fees | Chair | Member |
Board | A$360,000 | A$180,000 |
Audit and Risk Committee | A$36,000 | A$18,000 |
People Committee | A$36,000 | A$18,000 |
Non-Australian based NEDs continued to be paid in 2025 in line with the exchange rate at the time of their appointment.
5.2.1. 2025 equity disclosures
The following equity vested to NEDs during 2025:
Equity type | Grant details | Vesting details | Exercise details | ||||
Date | # units | Date | Exercise price | Date | Resultant shares | Value exercised | |
2022 NED SARs | 18-May-22 | 52,070 | 18-May-25 | A$4.95 | n/a | n/a | 0 |
1 Following Shareholder approval, premium-priced unlisted share options were issued to Ms. Olson in 2022. The amounts recorded for share based payments (options) for NEDs reflect the fair value of these options expensed each year over the life of the option.
2 Ms. McDonald joined the Board on March 3, 2025, and her remuneration for 2025 is for the part year worked.
3 Ms. Olson resigned as Chair and Non-Executive Director, effective February 3, 2026. Ms. Olson was paid in USD with an exchange rate as at the date he joined the Board of 0.7527.
4 Mr. McCann retired from the Board on May 21, 2025. His remuneration reported for 2025 is for the part year worked. The non-cash benefit provided related to a retirement gift, funded between the Directors and Telix.
5 No superannuation was applicable for Dr. Kluge in 2024 as he did not provide services in Australia. Ms. Olson has a certificate of coverage, which exempts the Group from paying superannuation.
Dr. Kluge was paid in Euro (€) with an exchange rate as at the date he joined the Board of 0.69326. Dr. Kluge retired from the Board on October 17, 2024, and his remuneration reported for 2024 is for the part year worked
190
5.3. 2025 NED statutory remuneration
The table below sets out NED remuneration for 2025 and 2024, prepared in accordance with relevant IFRS and Australian Accounting Standards.
All numbers have been restated for 2024 and provided in 2025 in US$ using the average exchange rate over the 2025 year.
Directors' Fees | Superannuation | Share-based payment1 | Non-cash benefits | Total | Options | |||
Name | Year | US$ | US$ | US$ | US$ | US$ | US$ | % |
NEDs | ||||||||
Ms. McDonald2 | 2025 | 104,332 | 12,314 | - | - | 116,646 | - | - |
2024 | - | - | - | - | - | - | - | |
Mr. Nelson | 2025 | 115,898 | 13,618 | - | - | 129,516 | - | - |
2024 | 80,164 | 9,018 | - | - | 89,182 | - | - | |
Ms. Skinner | 2025 | 134,971 | 15,864 | - | - | 150,835 | - | - |
2024 | 92,071 | 10,357 | - | - | 102,428 | - | - | |
Former NEDs | ||||||||
Ms. Olson3 | 2025 | 230,380 | - | - | - | 230,380 | - | - |
2024 | 97,211 | - | 37,730 | - | 134,942 | 37,730 | 27.96 | |
Mr. McCann4 | 2025 | 88,296 | 9,398 | - | 19,628 | 117,322 | - | - |
2024 | 148,465 | 16,701 | - | - | 165,166 | - | - | |
Dr. Kluge5 | 2025 | - | - | - | - | - | - | - |
2024 | 68,779 | - | - | - | 68,779 | - | - | |
Total | 2025 | 673,877 | 51,194 | - | 19,628 | 744,699 | - | n/a |
2024 | 486,690 | 36,076 | 37,730 | - | 560,497 | 37,730 | n/a |
191
5.4. 2026 NED remuneration
As previously disclosed, Telix experienced significant growth in market capitalization, revenue and headcount in
2024 and into 2025. As a result of this significant growth, and similar to the position for Executive KMP, Mercer
was also requested to provide updated remuneration benchmarking for the NED group. The same global peer
group that was used for Executive KMP was also used for NED benchmarking.
In addition, an ASX peer group was used comprising 20 companies in the health care, communication services,
information technology and industrials (production/manufacturing only) sectors, with Telix situated in the middle by
market capitalization. Regard was also had to a further group of 14 ASX listed companies, with international operations
and NEDs resident overseas.
The benchmarking data indicated that NED Board and Committee fees were generally below benchmark, in some cases
significantly so. Having regard to the shareholder experience in 2025, the Board does not presently propose to increase
NED fees for 2026. However, the Board is undertaking a search for internationally experienced directors and if it is
necessary to adjust the fees (including through the addition of equity, subject to shareholder approval) in order to attract
high calibre directors, an adjustment to fees may be considered.
6. Remuneration governance
6.1 Governance framework
The governance of Telix’s remuneration framework ensures:
•The Board delegates specific responsibilities to the People Committee to provide recommendations to the Board
•Telix’s strategic objectives, corporate governance principles, market practice and stakeholder interests are
considered, and
•Achievement of pre-determined financial results and strategic objectives is rewarded through sustainable
means.
Roles in the Governance framework | |
THE BOARD has overall responsibility for oversight of Telix’s remuneration approach for KMP (NEDs and Executives), including: •evaluating performance, determining remuneration outcomes and succession planning for the MD & CEO •determining remuneration outcomes, monitoring performance and succession planning of NEDs and Other Executive KMP, and •approving the Group’s remuneration policies and practices. | THE PEOPLE COMMITTEE assists the Board in fulfilling its responsibilities to shareholders and regulators in relation to the Group’s people and culture, remuneration and workplace health and safety policies and practices, including: •Telix’s remuneration framework and policies, including Telix’s Equity Incentive Plan rules; •remuneration arrangements and outcomes for KMP (NEDs and Other Executive KMP), including in respect of short term and long term variable remuneration, •remuneration related reporting and disclosures. The People Committee may engage external advisors to provide information to assist in making remuneration decisions. |
MANAGEMENT provides relevant information and analysis required to support effective decision making, including for remuneration related considerations. | EXTERNAL ADVISORS may be engaged by the People Committee to provide: •information to support effective decision making •an external perspective to assist in analysis with their expertize for remuneration related matters, and •on occasion, to provide remuneration recommendation/s as defined by section 9B of the Australian Corporations Act 2001 (Cth). |
AUDIT AND RISK COMMITTEE assists the Board with the Group’s risk management framework and risk appetite. | |
As detailed in sections 4.2 and 5.6, during 2025, Mercer was engaged to provide market benchmarking data for
Executive KMP and NEDs. They provided data only, and did not provide any remuneration recommendations in 2025.
Further information on the Board’s role and Telix’s corporate governance policies (including the Securities Dealing Policy)
is available in Telix’s 2025 Corporate Governance Statement and on Telix’s website at: ir.telixpharma.com/governance/
documents/charters. Telix’s Securities Dealing Policy prohibits hedging or margin lending in respect of Telix securities.
192
6.2. Malus and clawback
The Board in its sole discretion, may reduce, cancel in full, or seek to clawback any incentive provided to any Executive
KMP, including former Executive KMP, if it determines that at any time the Executive KMP:
•Acted dishonestly (including, but not limited to, misappropriating funds or deliberately concealing a transaction)
•Acted or failed to act in a way that contributed to Telix making a material financial misstatement including where
Telix is required to prepare an 'Accounting Restatement' for the purposes of Telix's Clawback / Dodd-Frank
Compensation Recovery Policy
•Acted or failed to act in a way that contributed to a breach of a significant legal or regulatory requirement
•Acted or failed to act in a way that contributed to Telix incurring significant reputational harm, a significant
unexpected financial loss, impairment charge, cost or provision
•Exposed employees, the broader community or environment to excessive risks, including health and safety
•Breached their post-employment restraints (unless otherwise determined by the Board)
•Committed a material breach or non-compliance with Telix’s Code of Conduct and/or any other employee or
governance related policies, and/or
•Took excessive material risks or contributed to or may benefit from unacceptable cultures within the Group.
During 2025, the Board exercised no malus or clawback.
193
6.3. Equity Incentive Plan
The purpose of the EIP is to align employees’ and directors’ interests with shareholders’ interests by providing equity as part of remuneration arrangements.
The EIP enables our Board of Directors to award different types of equity instruments tailored to specific applications. These can include rights to acquire shares contingent on
meeting specified performance metrics, options to acquire shares on payment of an exercise price and rights and/or options that are contingent on remaining in employment,
among others. We offer three types of securities under the EIP, including share options, share rights (including share appreciation rights) and restricted shares, which we refer to
as Incentive Securities.
Feature | Details | |
Eligibility | The Board determines the full-time or part-time employees (including a Director employed in an executive capacity), NEDs, casual employees or contractors or any other eligible persons (determined at the Board’s discretion) that may participate in the EIP, collectively referred to as Eligible Employees. Casual and contractor staff must be employed on at least a 40% Full Time Equivalent (FTE) to participate in line with the EIP rules. | |
Administration of the EIP | The EIP is administered by our board of directors, who have the power to determine the appropriate procedures for the EIP. | |
Invitation | The Board of Directors may make an invitation to an Eligible Employee to apply for Incentive Securities on such terms and conditions as the board of directors determines from time-to-time, including with relation to the Incentive Securities: (1)the type and number and/or the method by which the number will be calculated; (2)the amount (if any) payable for the grant; (3)any vesting conditions or other conditions; (4)the procedure for exercising an option or right following vesting; (5)the determination the board of directors has made at its discretion that vesting of share rights and/or exercise of options (as applicable) will be satisfied through an allocation of shares or by cash payment; (6)the circumstances in which rights and/or options will lapse, shares allocated under the EIP may be forfeited or an EIP participant’s entitlement may be reduced/extinguished; (7)how the securities may be treated in the event that an Eligible Employee ceases employment; (8)any restrictions on dealing shares; and (9)any other terms and conditions that, in the opinion of our board of directors, are fair and reasonable and not inconsistent with the EIP, and any other information that is required by applicable law. | |
Grant price | Unless the board of directors determines otherwise, no payment is required for the grant of Incentive Securities under the EIP. | |
Cap on number of ordinary shares to be issued under the EIP | The number of equity securities offered to participants under the EIP must not, when aggregated with the number of equity securities issued over the prior three years under (i) the EIP; (ii) any other employee share scheme covered by the ASIC Instrument 2022/1021; or (iii) an ASIC-exempt arrangement of a similar kind to an employee incentive scheme, exceed 32,405,821 equity securities, as approved by shareholders at an annual general meeting of shareholders on May 22, 2024. Our board of directors retains the discretion to adjust the cap on the number of the shares to be issued under the EIP, so long as the adjustment complies with applicable law. | |
Rights attaching to shares (including restricted shares) | Ranking. Shares issued under the EIP rank equally with other fully paid ordinary shares at the time of issue, except in relation to any rights attaching to such shares by reference to a record date prior to the date of their issue. Dividends. Holders of shares granted under the EIP are entitled to participate in all dividends and other distributions or benefits payable to participants in respect of their shares. Voting rights. Holders of shares granted under the EIP are entitled to exercise all voting rights attached to their shares, either generally or in a particular case, in accordance with our Constitution. | |
194
Feature | Details | |
Options | Exercise price | The board of directors shall advise each Eligible Employee in the offer documentation the procedure for exercising share options, including any exercise price that will become payable with respect to the share options exercised. Subject to ASX listing rules, prior to the exercise of share options, the board of directors will retain the power to adjust the relevant exercise price in order to minimize or eliminate any material advantage or disadvantage to a participant resulting from a corporate action by, or capital reconstruction in relation to, the Company. |
Exercise period | Share options will vest and become exercisable when all vesting conditions and any other conditions advised to the participant by the board of directors have been tested and satisfied (or otherwise waived by the board of directors). If the vesting conditions and all other relevant conditions are satisfied during a period in which the participant is prohibited from dealing in our securities or shares, the board of directors may determine that the vesting of the options held by the affected participant will be delayed until such dealings are permitted. | |
Lapse of share options | Share options will lapse upon the earliest to occur of: (1)ten years after the date on which the options were allocated to the participant, or any other date nominated as the expiry date of the offer; (2)the option lapsing in accordance with a provision of the EIP; (3)failure to meet a vesting condition or any other applicable condition within the vesting period; or (4)our receipt of a written notice from the participant that the participant has elected to surrender the option. | |
Shares issued | Upon the exercise of a share option, we will issue the number of fully paid ordinary shares allocatable to the share options that have been exercised, ranking equally with, and having the same rights and entitlements as, other ordinary shares on issue at the date of allotment of the share (other than rights and entitlements accrued prior to the date of allotment of the share). Notwithstanding, the board of directors may determine that the exercise of an option will be satisfied in part or in whole by a cash payment in lieu of an allocation of shares. | |
Restrictions on transfer of share options | In the case of options held by/on behalf of a participant who is a director, vested options must be satisfied by shares that have been purchased on market, unless (1)no shareholder approval is required under the listing rules in respect of the director’s participation in the EIP; or (2)shareholder approval has been obtained for the director’s participation in the EIP to the extent required under the listing rules. | |
Share Rights | Exercise price | No amount is payable with respect to share rights upon vesting and exercise. |
Exercise period | Share rights will vest and become exercisable (or will automatically be exercised, if specified by the board of directors in the terms provided at the time of grant) when all vesting conditions and any other conditions advised to the participant by the board of directors have been satisfied (or otherwise waived by the board of directors). If the vesting conditions and all other relevant conditions are satisfied during a period in which the participant is prohibited from dealing in our securities or shares, the Board may determine that the vesting of the rights held by the affected participant will be delayed until such dealings are permitted. | |
Lapse of share rights | The share rights will lapse upon the earliest to occur of: (1)ten years after the date the rights were allocated to the participant, or any other date nominated as the expiry date in the offer; (2)the rights lapsing in accordance with a provision of the EIP; (3)failure to meet a vesting condition or any other applicable condition within the vesting period; or (4)receipt of a written notice from the participant that the participant has elected to surrender the right. | |
Shares issued | Upon vesting, the board of directors will issue the number of fully paid ordinary shares allocatable to the share rights that have vested, ranking equally with, and having the same rights and entitlements as, our other ordinary shares on issue at the date of allotment of the share (other than rights and entitlements accrued prior to the date of allotment of the share). Notwithstanding, the board of directors may determine that the exercise of a share right will be satisfied in part or in whole by a cash payment made in lieu of an allocation of shares. In the case of share rights held by a participant who is a director, vested rights must be satisfied by shares that have been purchased on market, unless: (1)no shareholder approval is required under the listing rules in respect of the director’s participation in the EIP; or (2)shareholder approval has been obtained for the director’s participation in the EIP to the extent required under the listing rules. | |
195
Feature | Details | |
Share appreciation rights | At its discretion, the board of directors may determine that share appreciation rights will be granted to Eligible Employees. Share appreciation rights are share rights which only produce value if, at the time of vesting and exercise, the current market price exceeds a notional price specified by the board of directors at the time of the offer of such share appreciation rights. In the event that the calculation of current market price less notional price results in a zero or negative value at the time of exercise, the participant will not be entitled to any issuance of shares or cash payment. In the event that such calculation returns a positive value, the participant will be entitled to shares (or cash payment, as determined by the board of directors under the applicable rules of the EIP) with a value equal to the excess of the current market value over the notional price. Notwithstanding, the remainder of the terms of the EIP applicable to share rights (including exercise period, lapse, and restrictions on transfer) apply equally to share appreciation rights. | |
Restrictions on transfer of share rights | Unless the board of directors determines otherwise, share rights may not be registered in any name other than that of the participant and may not be transferred, assigned, or otherwise dealt with by the participant. | |
Restricted Shares | Cessation of restrictions | A restricted share ceases to be restricted (i.e., vests) where the vesting period and all other relevant conditions have been satisfied or waived by the board of directors and the participant has been notified that the restrictions have ceased or no longer apply. If the vesting conditions and all other relevant conditions are satisfied during a period in which the participant is prohibited from dealing in our securities or shares, the board of directors may determine that the vesting of the restricted shares held by the affected participant will be delayed until such dealings are permitted. |
Forfeiture of restricted shares | A restricted share will be forfeited upon the earliest to occur of: (1)the restricted share being forfeited in accordance with a provision of the EIP; (2)the failure to meet a vesting condition or other applicable condition within the vesting period; or (3)our receipt of a written notice from the participant that the participant has elected to surrender the restricted share. | |
Vesting conditions | Incentive Securities may be subject to any vesting condition the board of directors determines. Incentive Securities will vest to the participant upon all the vesting conditions and any other applicable conditions that apply to such Incentive Securities being satisfied. The board of directors has discretion to attach individual vesting conditions to the Incentive Securities at the time they are issued. Eligible Employees will be advised of such vesting conditions in connection with their invitation to participate in a grant. The board of directors may in its absolute discretion waive, amend, or replace any or all of the vesting conditions, provided that the interests of the affected participant are not, in the opinion of the Board, materially prejudiced or advantaged relative to the position reasonably anticipated at the time of grant. | |
Employee Share Purchase Plan Shares ("ESPP Shares") | For Australian participants only, ESPP Shares will be allocated via new issue twice per year: in September and the following March, based on payroll deductions over the calendar year. The September purchase will include salary deductions between January 1 and June 30, and the March purchase based on salary deductions between July 1 and December 31. These will be held as Restricted Shares under the EIP, until they pass an approximately 18-month restricted period, at which point they will be released to the participant (in an open trading window). For US participants, please see the section below “U.S. Employee Stock Purchase Plan.” | |
Amendments, suspensions or termination to/of the EIP | Subject to the exceptions listed below, our board of directors may at any time by resolution amend, suspend or terminate any provision of the EIP without the consent of the participant. However, no amendment, suspension or termination may be made if the amendment, suspension or termination materially prejudices the rights of any participant as they existed before the date of the relevant amendment, suspension or termination. The exceptions are amendments introduced: (1)for complying or conforming with present or future laws or regulations; (2)to correct any manifest error or mistake; or (3)to take into consideration adverse taxation implications in relation to the EIP. Moreover, the board of directors may waive, amend or replace any vesting condition attaching to an Incentive Security if the board of directors determines that the original vesting condition is no longer appropriate or applicable. | |
196
6.4. U.S. Employee Stock Purchase Plan
On December 12, 2024, the Board approved and adopted the Telix Pharmaceuticals (US) Inc. Employee Stock Purchase
Plan Rules ("U.S. ESPP"), subject to shareholder approval. Shareholders subsequently approved the U.S. ESPP on May 21,
2025 at the 2025 AGM.
The material terms of the U.S. ESPP are summarized below.
General
The U.S. ESPP is comprised of two distinct components in order to provide flexibility to grant options to purchase shares
under the U.S. ESPP. Specifically, the U.S. ESPP authorizes (1) the grant of options to U.S. employees that are intended
to qualify for favorable U.S. federal tax treatment under Section 423 of the Internal Revenue Code of 1986, as amended
("the Code") referred to as the Section 423 Component, and (2) the grant of options that are not intended to be tax-
qualified under Section 423 of the Code to provide flexibility to achieve tax, securities laws or other objectives, which are
referred to as the Non-Section 423 Component. Where permitted under local law and custom, we expect that the Non-
Section 423 Component will generally be operated and administered on terms and conditions similar to the Section 423
Component. In order for the Section 423 Component to qualify for U.S. federal tax treatment under Section 423 of the
Code, shareholders must approve the U.S. ESPP within 12 months of adoption by the Board.
Shares Available for Awards; Administration
A total of 1,351,000 ordinary shares are initially reserved for issuance under the U.S. ESPP. The board of directors, or a
committee of the board of directors, will be the administrator of the U.S. ESPP, or the Administrator, and has authority to
interpret the terms of the U.S. ESPP and determine eligibility of participants. The People Committee is the initial
Administrator of the U.S. ESPP. The ordinary shares purchased under the U.S. ESPP may be authorized but unissued
shares or treasury shares, including shares bought on the open market, new issue, recycled, or otherwise acquired for
purposes of the U.S. ESPP. The Administrator will have the authority to grant options to purchase ADSs to eligible
participants in lieu of ordinary shares.
Eligibility
U.S. employees who have been continuously employed by a participating company within the Group (as determined by
the Administrator) for at least six months prior to the enrollment date, and whose customary employment is 20 hours or
more per week, are eligible to participate in the U.S. ESPP. However, an employee may not be granted an option to
purchase shares under the U.S. ESPP if the employee, immediately after the grant, would own (directly or through
attribution) shares possessing 5% or more of the total combined voting power or value of all classes of Telix shares or
shares of any of Telix's subsidiaries.
Offering Periods and Purchase Price
Shares will be offered under the U.S. ESPP during offering periods. Eligible participants will be able to enroll in the U.S.
ESPP during a specified enrollment period each calendar year, with each plan year commencing on January 1 (other than
the first plan year for calendar year 2025, which commenced on April 1, 2025). Starting in calendar year 2026, there will
be a series of sequential offering periods, with the first offering period commencing on or around January 1 of each
calendar year and ending on or before September 30 of that same year, and with the second offering period
commencing on or around July 1 and ending on or before March 15 of the following calendar year. Within each offering
period, there will be a six month contribution period, starting on January 1 and July 1 of each calendar year, respectively,
within which employee payroll deductions will be collected from participants and accumulated under the plan for the
applicable offering period. If permitted by the Administrator to comply with non-U.S. requirements for the Non-423
Component, in lieu of payroll deductions, the Administrator may provide that an eligible employee may elect to
participate via cash, check or other means acceptable to the Administrator. Employee contributions will be used to
allocate shares (via purchase or new issue of shares) on each exercise date during an offering period. The exercise dates
for each offering period will be the final trading day in the offering period. The Administrator may, in its discretion, modify
the terms of future offering and contribution periods.
The U.S. ESPP permits participants to have contributions made during the contribution periods to be expressed, at Telix's
sole discretion, as a U.S. dollar amount with the minimum and/or maximum U.S. dollar amount to be specified by Telix.
These contributions will be used to purchase ordinary shares. The Administrator may establish a minimum or maximum
amount of contributions a participant may make, and the minimum or maximum number of shares that may be purchased
during any offering period. In addition, no employee will be permitted to accrue an option to purchase shares under the
Section 423 Component at a rate in excess of US$25,000 worth of shares during any calendar year during which such a
purchase right is outstanding (determined at the fair market value per share of our ordinary shares as of the beginning of
each offering period).
On the first trading day of each offering period, each participant will automatically be granted an option to purchase
ordinary shares. An option will be exercised automatically at the end of the applicable offering period to the extent of the
payroll deductions accumulated during the offering period. The purchase price of the shares will be 85% (or such greater
percentage, if determined by the Administrator prior to the offering period) of the lower of the fair market value of Telix
ordinary shares on the first trading day of the offering period or on the exercise date. The fair market value of Telix's
ordinary shares for this purpose will generally be the closing price of our ordinary shares on the ASX (or such other
197
exchange as our ordinary shares may be traded at the relevant time, including our ADSs on Nasdaq) on the first trading
day of the offering period or on the exercise date, as applicable.
Participant Modification, Withdrawal or Termination
Participants may voluntarily end their participation in the U.S. ESPP at any time during a contribution period prior to the
end of the applicable offering period, and will be paid their accrued payroll deductions that have not yet been used to
purchase ordinary shares. Participants will not be permitted to modify their plan year contributions following close of the
specified enrollment period. Participation ends automatically upon a participant’s termination of employment. A
participant’s contributions will roll over in each new plan year until termination of participation, or modification or
discontinuance of contributions consistent with the terms of the U.S. ESPP.
Transfer of Rights
A participant may not transfer, assign, pledge or otherwise dispose of contributions made or rights granted under the
U.S. ESPP other than in the event of the death of a participant, in which case such participant’s contributions will be
delivered to the executor or administrator of the estate of the participant. The Administrator will have the discretion at its
election to impose a holding period during which the sale of shares issued under the U.S. ESPP will be restricted. During
the initial offering period, the terms of the U.S. ESPP require the Administrator to impose an 18-month holding period
from each exercise date, during which the sale of shares issued under the U.S. ESPP will be restricted from transfer.
Except for the aforementioned transfer restrictions, once shares have been delivered to participants, such participants
will have all of the rights and privileges applicable to other holders of ordinary shares.
Changes to Shares or Capital Structure
Subject to any required shareholder approval, in the event that there is a specified type of change in our capital
structure, such as a share split, share dividend, or combination or reclassification of the shares, proportionate
adjustments will be made to the number of shares reserved under the U.S. ESPP, the price of shares that any participant
has elected to purchase, and any other variables tied to the number of shares or the per share purchase price which we
determine should be adjusted.
Amendment and Termination
The Administrator may amend, suspend or terminate the U.S. ESPP at any time without the consent of the participant.
However, to the extent that shareholder approval is required under applicable tax, securities, stock exchange or other
laws or rules, no amendment may be made without such approval.
1
2
198
7. Additional statutory disclosures
7.1. Ordinary shareholdings
The relevant interests of KMP in the shares issued by Telix, held directly, indirectly or beneficially either personally or by their related parties are included in this section.
7.1.1. NED ordinary shareholdings
Name | Balance January 1, 2025 | Shares issued from Options exercised | Net acquired/ (disposed) | Other changes | Balance December 31, 2025 |
Mr. McCann1 | 1,150,000 | - | (30,000) | (1,120,000) | - |
Ms. Olson2 | 95,235 | - | 11,315 | - | 106,550 |
Mr. Nelson | 3,628,750 | - | - | - | 3,628,750 |
Ms. McDonald | - | - | 3,719 | - | 3,719 |
Ms. Skinner | 595,000 | - | - | - | 595,000 |
5,468,985 | - | (14,966) | (1,120,000) | 4,334,019 |
1 Mr. McCann's ordinary shareholdings are reflected as Nil at year end as he is no longer a KMP
2 Ms. Olson resigned as Chair and Non-Executive Director, effective February 3, 2026
7.1.2. Executive KMP ordinary shareholdings
Name | Balance January 1, 2025 | Shares issued from Options exercised | Net acquired/ (disposed) | Other changes | Balance December 31, 2025 | % of base salary held in shares1 |
Dr. Behrenbruch | 23,228,298 | 100,708 | (2,000,000) | - | 21,329,006 | 29,895% |
Mr. Smith | 6,500 | 1,096 | - | - | 7,596 | 12% |
Dr. Cade | 373,133 | - | - | - | 373,133 | 775% |
Dr. Patti | - | 145,000 | (119,442) | - | 25,558 | 45% |
23,607,931 | 246,804 | (2,119,442) | - | 21,735,293 |
1 As at December 31, 2025, the Executive KMP’s shareholding as a percentage of base salary is calculated using the closing share price of A$11.20 on the ASX.
1 Ms. Olson resigned as Chair and Non-Executive Director, effective February 3, 2026
199
7.2. KMP option holdings for the year ended December 31, 2025
Equity-based awards granted to our non-executive directors and executive officers consist of options (no longer issued) and PSARs that provide the holder with the right to
convert each option or PSAR to a fully paid ordinary share if vesting conditions are met. The following table discloses particulars of all awards outstanding for non-executive
directors and executive officers as of December 31, 2025, including awards (as options) granted before fiscal year 2025. All values in A$, except where noted.
7.2.1. NED option holdings
Name | Grant date of options | Number of options granted | Exercise price | Expiry date | Fair value per option at grant date | Vesting date | Vesting number | Vested during the year | Lapsed or forfeited during the year | Exercised in current or prior year | Eligible to exercise at December 31, 2025 | Unvested at December 31, 2025 | Maximum value yet to vest |
Ms. Olson1 | 18-May-22 | 52,070 | A$4.95 | 18-May-26 | A$2.19 | 31-Dec-24 | 52,070 | 52,070 | - | - | 52,070 | - | - |
Total | 52,070 | 52,070 | 52,070 | - | - | - | - | - |
200
7.2.2. Executive KMP option holdings
Name | Grant date of options | Number of options granted | Exercise price A$ / US$ | Expiry date | Fair value per option at grant date A$ / US$ | Vesting date | Vesting number | Vested during the year | Lapsed or forfeited during the year | Exercised in current or prior year | Eligible to exercise at December 31, 2025 | Unvested at December 31, 2025 | Maximum value yet to vest US$ |
Dr. Behrenbruch | 12-Jan-20 | 200,000 | A$2.23 | 12-Jan-24 | A$0.46 | 12-Jan-23 | 200,000 | 200,000 | - | (200,000) | - | - | - |
26-Jan-21 | 100,708 | A$4.38 | 26-Jan-26 | A$2.12 | 28-Oct-22 | 100,708 | 100,708 | - | (100,708) | - | - | - | |
05-Apr-22 | 139,672 | A$4.95 | 04-Apr-27 | A$2.43 | 31-Dec-24 | 139,672 | 139,672 | (46,558) | - | - | 93,114 | 75,645 | |
30-May-23 | 120,268 | A$6.90 | 31-Dec-27 | A$7.65 | 31-Dec-25 | 120,268 | - | - | - | - | 120,268 | 102,741 | |
26-Jun-24 | 144,037 | A$11.94 | 31-Mar-29 | A$8.57 | 31-Mar-27 | 144,037 | - | - | - | - | 144,037 | 427,881 | |
21-May-25 | 166,483 | A$28.67 | 31-Mar-30 | A$10.81 | 31-Mar-28 | 166,483 | - | - | - | - | 166,483 | 1,141,166 | |
21-May-25 | 2,595 | A$0.00 | 31-Mar-28 | A$25.68 | 28-Feb-26 | 2,595 | - | - | - | - | 2,595 | 9,310 | |
Mr. Smith | 24-Oct-22 | 45,449 | A$6.15 | 24-Oct-27 | A$3.08 | 24-Oct-25 | 45,449 | 45,449 | - | - | - | 45,449 | - |
24-Oct-22 | 32,463 | A$6.15 | 24-Oct-27 | A$3.08 | 24-Oct-25 | 32,463 | 32,463 | - | - | - | 32,463 | - | |
25-Apr-23 | 106,197 | A$6.90 | 31-Dec-27 | A$3.79 | 31-Dec-25 | 106,197 | - | - | - | - | 106,197 | 45,146 | |
25-Mar-24 | 35,000 | A$0.00 | 28-Feb-29 | A$11.70 | 31-Mar-27 | 35,000 | - | - | - | - | 35,000 | 198,312 | |
25-Mar-24 | 35,000 | A$0.00 | 28-Feb-30 | A$11.70 | 31-Mar-28 | 35,000 | - | - | - | - | 35,000 | 216,259 | |
13-May-24 | 76,311 | A$11.94 | 31-Mar-29 | A$7.59 | 31-Mar-27 | 76,311 | - | - | - | - | 76,311 | 263,252 | |
01-Jan-25 | 98,003 | A$28.67 | 31-Mar-30 | A$14.09 | 31-Mar-28 | 98,003 | - | - | - | - | 98,003 | 854,148 | |
01-Jan-25 | 1,111 | A$0.00 | 31-Mar-28 | A$25.56 | 28-Feb-26 | 1,111 | - | - | - | - | 1,111 | 2,655 | |
17-Mar-25 | 1,096 | A$12.16 | 30-Sep-25 | A$15.70 | 01-Sep-25 | 1,096 | 1,096 | - | (1,096) | - | - | - | |
17-Mar-25 | 893 | A$22.40 | 15-Mar-26 | A$7.70 | 01-Mar-26 | 893 | - | - | - | - | 893 | 795 | |
Dr. Cade | 04-Nov-19 | 500,000 | A$2.30 | 04- Nov-23 | A$0.48 | 04-Nov-22 | 500,000 | 500,000 | - | (500,000) | - | - | 637 |
26-Jan-21 | 85,347 | A$4.38 | 26-Jan-26 | A$2.12 | 28-Oct-22 | 85,347 | 85,347 | - | (85,347) | - | - | - | |
21-Jul-21 | 100,000 | A$0.00 | 20-Jul-26 | A$5.35 | 20-Jul-26 | 100,000 | - | - | - | - | 100,000 | 358,450 | |
05-Apr-22 | 78,189 | A$4.95 | 04-Apr-27 | A$2.43 | 31-Dec-24 | 78,189 | 78,189 | - | - | - | 78,189 | - | |
25-Apr-23 | 101,152 | A$6.90 | 31-Dec-27 | A$3.79 | 31-Dec-25 | 101,152 | - | - | - | - | 101,152 | 43,001 | |
13-May-24 | 74,191 | A$11.94 | 31-Mar-29 | A$7.59 | 31-Mar-27 | 74,191 | - | - | - | - | 74,191 | 255,937 | |
01-Jan-25 | 74,864 | A$28.67 | 31-Mar-30 | A$14.09 | 31-Mar-28 | 74,864 | - | - | - | - | 74,864 | 652,480 | |
01-Jan-25 | 1,080 | A$0.00 | 31-Mar-28 | A$25.56 | 28-Feb-26 | 1,080 | - | - | - | - | 1,080 | 2,580 |
201
Name | Grant date of options | Number of options granted | Exercise price A$ / US$ | Expiry date | Fair value per option at grant date A$ / US$ | Vesting date | Vesting number | Vested during the year | Lapsed or forfeited during the year | Exercised in current or prior year | Eligible to exercise at December 31, 2025 | Unvested at December 31, 2025 | Maximum value yet to vest US$ |
Dr. Patti | 21-Jul-21 | 100,000 | A$5.37 | 20-Jul-26 | A$2.62 | 28-Oct-22 | 100,000 | 100,000 | - | (100,000) | - | - | - |
05-Apr-22 | 15,826 | A$4.95 | 04-Apr-27 | A$2.43 | 31-Dec-24 | 15,826 | 15,826 | - | - | - | 15,826 | - | |
05-Apr-22 | 15,000 | A$0.00 | 04-Apr-27 | A$4.53 | 05-Apr-25 | 15,000 | 15,000 | - | (15,000) | - | - | - | |
02-May-23 | 32,938 | A$6.90 | 31-Dec-27 | A$3.79 | 31-Dec-25 | 32,938 | - | - | - | - | 32,938 | 14,002 | |
06-Jul-23 | 15,000 | A$0.00 | 31-Dec-25 | A$10.79 | 31-Dec-25 | 15,000 | - | - | (15,000) | - | - | - | |
01-Nov-23 | 15,000 | A$0.00 | 01-Nov-28 | A$8.99 | 31-Dec-26 | 15,000 | - | - | - | - | 15,000 | 39,632 | |
01-Nov-23 | 15,000 | A$0.00 | 01-Nov-29 | A$8.99 | 31-Dec-27 | 15,000 | - | - | - | - | 15,000 | 51,802 | |
13-May-24 | 17,175 | A$11.94 | 31-Mar-29 | A$7.59 | 31-Mar-27 | 17,175 | - | - | - | - | 17,175 | 59,249 | |
13-May-24 | 83,082 | A$11.94 | 31-Mar-29 | A$7.59 | 31-Mar-27 | 83,082 | - | - | - | - | 83,082 | 286,609 | |
26-Aug-24 | 15,000 | A$0.00 | 04-Apr-25 | A$19.86 | 01-Apr-25 | 15,000 | 15,000 | - | (15,000) | - | - | - | |
01-Jan-25 | 95,958 | US$19.99 | 31-Mar-30 | US$8.44 | 31-Mar-28 | 95,958 | - | - | - | - | 95,958 | 747,683 | |
01-Jan-25 | 1,230 | US$0.00 | 13-Mar-26 | US$17.43 | 27-Feb-26 | 1,230 | - | - | - | - | 1,230 | 2,945 | |
Total | 2,741,318 | 2,741,318 | 1,328,750 | (46,558) | (1,032,151) | - | 1,662,609 | 5,852,317 |
End of Remuneration Report
202
C.Board Practices
Director Terms
In accordance with the ASX Listing Rules, a director (other than the CEO) must not hold office, without re-election, past
the third annual general meeting ("AGM") following the director’s appointment or three years, whichever is longer. In
addition, under our Constitution, a director appointed by our Board who is not a CEO holds office until the next AGM of
the Company following his or her appointment and no director who is not the CEO may hold office without re-election
beyond the third AGM of the Company following the meeting at which such director was last elected (or re-elected).
Under our Constitution, to the extent that the ASX Listing Rules require an election of directors to be held and no director
would otherwise be required to submit for election or re-election, the director to retire is any director who wishes to
retire (whether or not he or she intends to stand for re-election), otherwise it is the director who has been longest in
office since their last election or appointment (excluding the CEO). As between directors who were last elected or
appointed on the same day, the director to retire must be decided by lot (unless they can agree among themselves).
Set forth below is information regarding the terms of service of each of our current directors:
Name | Title | Date of initial appointment | End of current term / Eligible for re-election |
Christian Behrenbruch | Managing Director and Group CEO | January 2017 | N/A |
Marie McDonald | Independent Non-Executive Director | March 2025 | 2028 AGM |
Mark Nelson | Interim Chair and Independent Non-Executive Director | September 2017 | 2026 AGM |
Jann Skinner | Independent Non-Executive Director | June 2018 | 2028 AGM |
Ms. Olson, our former Chair and Non-Executive Director, was appointed to our Board in March 2022, and served as Chair
of the Board from May 21, 2025 until her resignation, effective February 3, 2026.
Service Contracts
Other than as disclosed in this section, we do not have any service contracts with directors which provide for benefits
upon termination of employment.
Director Independence
As a foreign private issuer, under the listing requirements and rules of Nasdaq, we are not required to have independent
directors on our Board, except that our Audit and Risk Committee is required to consist fully of independent directors,
subject to certain phase-in schedules. However, our Board has determined that, under current listing requirements and
rules of Nasdaq and taking into account the Board's Charter independence requirements, Mark Nelson, Marie McDonald,
and Jann Skinner are “independent directors” under Rule 10A-3 of the Exchange Act, and pursuant to applicable Nasdaq
rules. In making such determination, our Board considered the relationships that each non-executive director has with us
and all other facts and circumstances our Board deemed relevant in determining the director’s independence, including
the number of ordinary shares beneficially owned by the director and his or her affiliated entities (if any).
The independence criteria under the applicable Nasdaq rules differ from the independence criteria set forth by the ASX
in the Corporate Governance Principles and Recommendations, 4th edition. Under the Corporate Governance Principles
and Recommendations, 4th edition, Mark Nelson, Marie McDonald, and Jann Skinner are “independent directors.”
Board Committees
To assist with the effective discharge of its duties, our Board has established an Audit and Risk Committee ("ARC"), a
People Committee, a Nomination Committee and a Disclosure Committee. Each committee (other than the Disclosure
Committee which reviews and approves all material announcements to the market, where not approved by the full Board
as specified by our Continuous Disclosure Policy and Disclosure Committee Charter) operates under a charter approved
by our Board, which sets forth the purposes and responsibilities of the committee as well as qualifications for committee
membership, committee structure and operations and committee reporting to our Board. Previously, the responsibilities
of the People Committee and the Nomination Committee were vested in the People, Culture, Nomination and
Remuneration Committee, which was dissolved effective March 3, 2025 in connection with the establishment of the
People Committee and the Nomination Committee. The primary roles of each of the committees, as set forth under their
respective Charters, is described below. Copies of the Charters are available under the Investor Centre section of our
website at www.telixpharma.com.
203
Audit and Risk Committee
The ARC has been established in accordance with our Constitution and operates under a Charter approved by our Board.
The ARC's role outlined in the Charter is to review and make recommendations (as appropriate) to our Board in relation to
its accounting, auditing, financial reporting, internal control, risk management, legal and regulatory compliance,
sustainability responsibilities (including related to climate change), and internal and external audit functions. The ARC has
also been delegated primary responsibility for overseeing the Group’s risk management processes (see the section titled
"Managing Risk - Role of the Board in Risk Oversight" below).
Under the Charter, the ARC is required to be comprised of at least three non-executive directors, all of whom must be
independent. In addition, none of the members may have participated in the preparation of the financial statements of
Telix or any current subsidiary at any time during the past three years.
The current membership of the ARC is:
•Jann Skinner (Chair);
•Marie McDonald; and
•Mark Nelson.
Ms. Olson, our former Chair and Non-Executive Director, served as a member of the ARC during the year ended
December 31, 2025, and until her resignation, effective February 3, 2026. Ms. McDonald was appointed as a member of
the ARC effective February 3, 2026.
People Committee
The People Committee has been established in accordance with our Constitution and operates under a Charter approved
by our Board. The People Committee’s roles outlined in the Charter include assisting our Board in fulfilling its
responsibilities relating to our key people and organizational culture strategies and their alignment with our purpose and
strategy, reviewing and making recommendations to the Board (as appropriate) with respect to Group remuneration,
including the Group's remuneration framework and policies, and the remuneration of Non-Executive Directors, the CEO
and other GET members (including KMP), approving the appointment and overseeing the succession planning of the
Group CFO and other KMP (other than the CEO), and the general terms of their employment contracts, oversight
responsibilities to shareholders with respect to our remuneration policies and practices, and belonging initiatives.
Under the Charter, the People Committee is required to be comprised of at least three non-executive directors, all of
whom must be independent.
The current membership of the People Committee is:
•Marie McDonald (Chair);
•Mark Nelson; and
•Jann Skinner.
Ms. Olson, our former Chair and Non-Executive Director, served as a member of the People Committee during the year
ended December 31, 2025, and until her resignation, effective February 3, 2026. Dr. Nelson was appointed as a member
of the People Committee effective February 3, 2026.
Nomination Committee
The Nomination Committee has been established in accordance with our Constitution and operates under a Charter
approved by our Board. The Nomination Committee's role outlined in the Charter is to assist the Board in the discharge of
its responsibilities related to, among other things, the size, composition and organization of the Board and its
committees, Board succession planning, including non-executive director appointments and appointment of the Group
CEO, assessing the independence of non-executive directors, the process for evaluating the performance of the Board
and its committees, the approval of the appointment and removal of the Group Company Secretary, and induction and
ongoing development for directors.
Under the Charter, the Nomination Committee is required to be comprised of at least three non-executive directors, all of
whom must be independent.
The current membership of the Nomination Committee is:
•Mark Nelson (Chair);
•Marie McDonald; and
204
•Jann Skinner.
Ms. Olson, our former Chair and Non-Executive Director, served as Chair of the Nomination Committee during the year
ended December 31, 2025, and until her resignation, effective February 3, 2026. Dr. Nelson was appointed Chair of the
Nomination Committee, and Ms. Skinner was appointed as a member of the Nomination Committee, effective February 3,
2026.
Managing Risk
Role of the Board in Risk Oversight
The ARC assists the Board in overseeing and reviewing the integrity of financial and corporate reporting, risk appetite
and the effectiveness of the enterprise risk management framework ("ERMF"), compliance systems and internal control
framework, the legality, propriety and materiality of related party transactions, and the external and internal audit
functions. In addition, the ARC has oversight of Telix’s sustainability strategy and reporting framework, including in
respect of climate change, healthcare and environmental governance and performance. The ARC receives reports from
management at least quarterly regarding our assessment of risks, having regard to the risk appetite approved by the
Board.
GET and other senior executives and internal and external auditors attend Committee meetings on invitation by the ARC.
The ARC holds regular meetings with the internal auditor, and separately with the external auditor without the
management team or MD & CEO present. Any Director who is not a member of the ARC may attend any meeting of the
Committee in an ex-officio capacity.
Risk Governance
The Company has adopted and follows an ERMF that incorporates the principles of effective risk management, as set out
in the Global Risk Management Standard ISO 31000, to identify, evaluate, monitor and manage risks in the Telix Group -
to improve business performance, remain innovative and establish competitive advantage, anticipate and communicate
uncertainties, reduce operational losses and surprises, and protect Telix’s reputation. Our ERMF data informs leaders in
their decision making from prioritizing activities, to resourcing, to escalation.
Risk and opportunity is managed through objective and consistent identification, assessment, monitoring, measurement
and reporting across the Telix Group. Management executes daily risk management activities, including by making
decisions within stated Board-delegated authority, ensuring employees understand their responsibilities for managing
risk through a “three lines” model, and establishing internal controls and guidance for the implementation of the ERMF.
In the “three lines” model, the first line, consisting of the business units and expert teams, executes core processes and
controls. The second line, comprising the Enterprise Risk Management function, sets policies, establishes frameworks to
manage risks, and delivers training and guidance to the first line. The third line, which constitutes internal and external
audit, provides independent review of the first and second lines.
Ultimate risk management oversight sits with the Board. The Board, with assistance from the ARC, sets the risk appetite,
within which it expects management to operate, and considers Telix’s risk profile on a regular basis to ensure it supports
the achievement of Telix’s strategic and corporate goals.
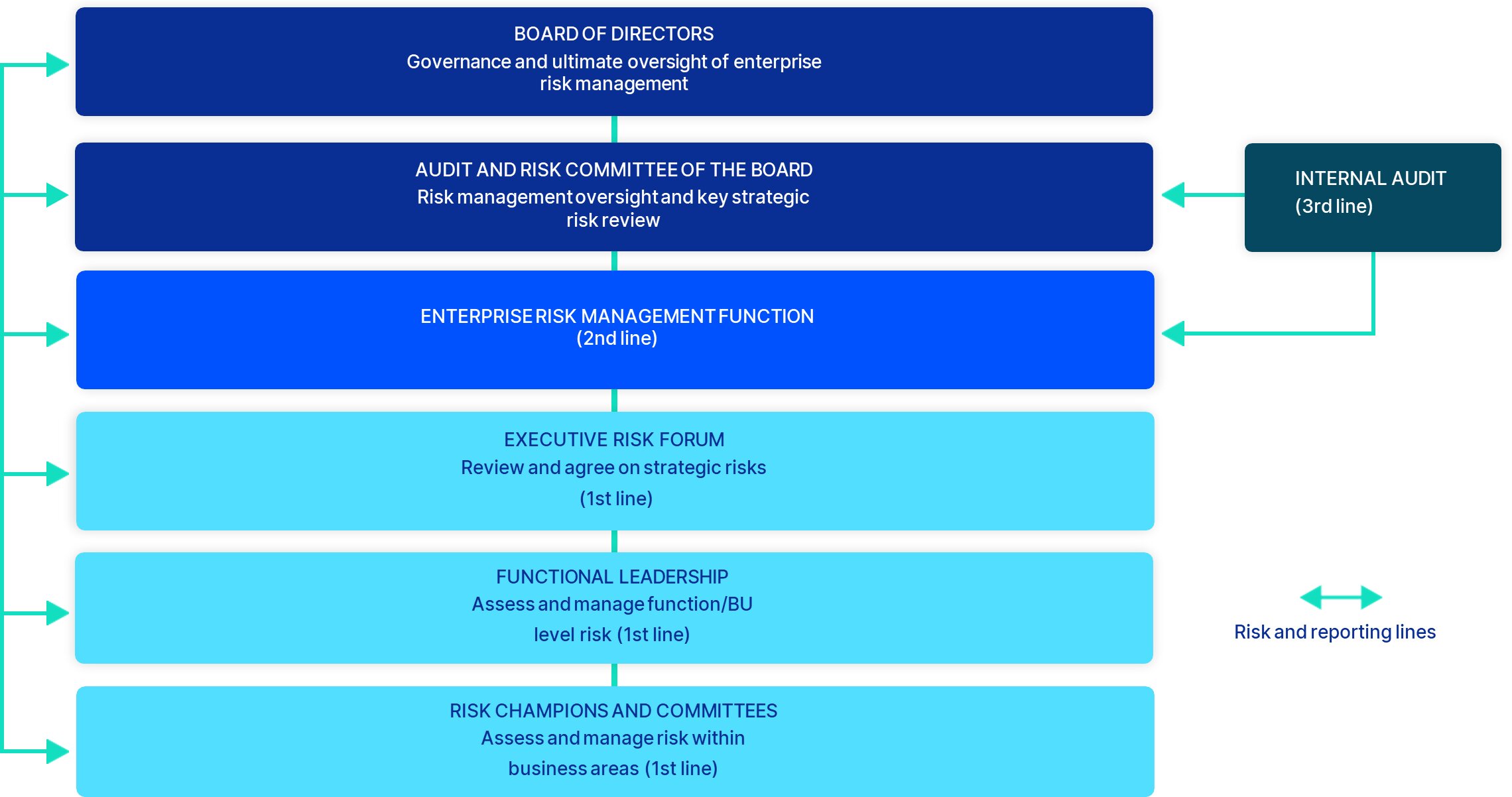
205
Foreign Private Issuer Exemption
We qualify as a “foreign private issuer” as defined in Rule 3b-4 of the Exchange Act. As a foreign private issuer, we are
exempt from certain rules under the Exchange Act that impose disclosure requirements as well as procedural
requirements for proxy solicitations under Section 14 of the Exchange Act. In addition, our executive officers and
directors are not subject to short-swing profit and insider trading reporting obligations under Section 16 of the Exchange
Act. They are, however, subject to the obligations to report changes in share ownership under Section 13 of the
Exchange Act and related SEC rules, to the extent applicable. Furthermore, absent an exemption from the SEC, and
effective March 18, 2026, our officers and directors will be subject to the insider reporting obligations under Section
16(a) of the Exchange Act, including the requirements to file Forms 3, 4 and 5, pursuant to the Holding Foreign Insiders
Accountable Act enacted on December 18, 2025. The foreign private issuer exemption also permits us to follow home
country corporate governance practices or requirements instead of certain Nasdaq listing requirements, including the
following:
•We rely on an exemption from the requirement that our independent directors meet regularly in executive sessions.
In contrast to Nasdaq requirements, the ASX Listing Rules and the Australian Corporations Act 2001 (Cth) do not
require the independent directors of an Australian company to have executive sessions.
•We rely on an exemption from the quorum requirements applicable to meetings of shareholders under Nasdaq rules.
While Nasdaq requires that an issuer provide for a quorum as specified in its bylaws for any meeting of the holders
of ordinary shares, which quorum may not be less than 33 1/3% of the outstanding shares of an issuer’s voting
ordinary shares, our Constitution provides that two shareholders present and entitled to vote on a resolution at the
meeting shall constitute a quorum for a general meeting.
•We rely on an exemption from the requirement that the responsibility for the appointment of the independent
registered public accounting firm be made by the audit committee. While the ARC is directly responsible for
remuneration and oversight of the independent registered public accounting firm, the ultimate responsibility for the
appointment of the independent registered public accounting firm rests with our shareholders in accordance with
Australian law and our Constitution. In accordance with Rule 10A-3 of the Exchange Act, the ARC is responsible for
the annual auditor engagement and if there is any proposed change to the independent registered public
accounting firm, the committee will make a recommendation to our board of directors, which would then be
considered by our shareholders at an annual meeting of shareholders.
•We rely on an exemption from the requirement prescribed by Nasdaq that issuers obtain shareholder approval prior
to the issuance of securities in connection with certain acquisitions, changes of control or private placements of
securities, or the establishment or amendment of certain stock option, purchase or other compensation plans.
Applicable Australian law and rules differ from Nasdaq requirements, with the ASX Listing Rules providing generally
for the ability to seek prior shareholder approval in numerous circumstances, including (i) issuance of equity
securities exceeding 15% of our issued share capital in any 12 month period (but, in determining the available issue
limit, securities issued under an exception to the rule or with shareholder approval are not counted), (ii) issuance of
equity securities to related parties, certain substantial shareholders and their respective associates (as defined in
the ASX Listing Rules) and (iii) directors or their associates acquiring securities under an employee incentive plan.
We intend to take all actions necessary for us to maintain compliance as a foreign private issuer under the applicable
corporate governance requirements of the Sarbanes-Oxley Act, the rules adopted by the SEC and the listing rules of
Nasdaq.
Australian Disclosure Requirements
Meetings and attendance
The Board meets as often as required. During the financial year ended December 31, 2025, the Board met 13 times.
Members of the Group Executive Team and other members of senior management attend meetings of the Board by
invitation. Each Board Committee provides a standing invitation for any Non-Executive Director to attend Committee
meetings (rather than just limiting attendance to Committee members). Committee agendas and papers are provided to
all Directors concerning matters to be considered. The table below excludes the attendance of Directors at Committee
meetings where they were not a Committee member.
1 The number of shares held refers to shares held either directly, indirectly or beneficially by Directors as at February 20, 2026. Where applicable, the information includes shares
held indirectly, including by nominee and/or other controlled entities
206
Board of Directors | Audit and Risk Committee | People Committee1 | Disclosure Committee | Nomination Committee | ||||||
Eligible to attend | Meetings attended | Eligible to attend | Meetings attended | Eligible to attend | Meetings attended | Eligible to attend | Meetings attended | Eligible to attend | Meetings attended | |
H K McCann2 | 5 | 5 | 3 | 3 | 2 | 2 | 2 | 2 | 1 | 1 |
T Olson | 13 | 13 | 5 | 5 | 4 | 4 | 3 | 3 | 3 | 3 |
C Behrenbruch3 | 13 | 13 | 3 | 3 | ||||||
M Nelson | 13 | 13 | 5 | 5 | 1 | 1 | 3 | 3 | ||
J Skinner | 13 | 12 | 5 | 5 | 4 | 4 | 2 | 2 | ||
M McDonald4 | 11 | 11 | 3 | 3 | 3 | 3 | ||||
A Whitaker5 | 1 | 1 | ||||||||
1.The People, Culture, Nomination and Remuneration Committee was dissolved and a separate People Committee of
the Board and Nomination Committee of the Board was established effective March 3, 2025. Attendance noted
combines the People, Culture, Nomination and Remuneration Committee and People Committee
2.H K McCann retired from the Board effective May 21, 2025
3.C Behrenbruch attends Audit and Risk Committee, People Committee and Nomination Committee meetings by
invitation
4.M McDonald was appointed Non-Executive Director, Chair of the People Committee and member of the Nomination
Committee effective March 3, 2025
5.A Whitaker was appointed Non-Executive Director effective April 7, 2025 and resigned April 29, 2025
Directors' interests in the securities of Telix
The relevant interests1 of each Director in the share capital of Telix as at the date of this report are included in Item 6
section 7.1.1.
Issue of unlisted equity securities
Unlisted ordinary shares under options or rights issued during the year were as follows:
Options/Rights granted | ASX code | Expiry date | Exercise price | Number under option |
TLXO030 | TLXAO | March 31, 2030 | A$28.67 | 1,700,510 |
TLXO031 | TLXAU | March 31, 2028 | nil | 10,561 |
TLXO032 | TLXAO | March 31, 2028 | A$14.90 | 742,992 |
TLXO033 | Not applicable | September 30, 2025 | A$12.16 | 14,353 |
TLXO034 | Not applicable | March 15, 2026 | A$22.40 | 11,815 |
TLXO030-ADS | TLXAW | March 31, 2030 | US$19.99 | 2,881,250 |
TLXO031-ADS | TLXAV | March 13, 2026 | nil | 10,342 |
TLXO032-ADS | TLXAW | March 31, 2028 | US$9.98 | 1,758,187 |
TLXO033-ADS | Not applicable | September 30, 2025 | US$8.02 | 26,361 |
TLXO034-ADS | Not applicable | March 15, 2026 | US$13.61 | 28,776 |
Performance rights | TLXAR | January 31, 2030 | nil | 3,914,631 |
Shares issued for acquisitions, on exercise of rights or options and lapse of options
Acquisition share issues and exercise of rights
The Company completed the following acquisitions during the period, which resulted in the respective issue of fully paid
ordinary shares and rights outlined below.
On January 31, 2025, Telix completed the acquisition of ImaginAb, Inc. The purchase price was paid to shareholders in
equity through the issue of 2,053,311 fully paid ordinary Telix shares and the balance paid in cash.
207
In addition to the above, milestone rights have been issued to ImaginAb shareholders. Each milestone has a fixed dollar
amount which can be settled, at Telix's election, either in cash or shares.
On January 31, 2025, March 17, 2025 and 6 May 2025, a total of 94,512, 83,395 and 91,168 shares were issued
respectively following the satisfaction and exercise of Lightpoint performance rights.
On March 17, 2025 13,356 and on October 29, 2025 24,219 shares were issued following the satisfaction and exercise of
Dedicaid performance rights.
Employee exercise or lapse of options
Ordinary shares of Telix issued during the financial year ended December 31, 2025 on the exercise of options granted
over unissued shares and lapse of options were as follows:
•a total of 1,692,603 fully paid ordinary shares were issued upon exercise of 2,272,000 unlisted share options or
PSARs, and
•a total of 1,850,000 share options lapsed unexercised. These options lapsed in accordance with the terms of
their grant.
Since December 31, 2025 to the date of this Annual Report, no shares were issued from the exercise of options under
Telix’s Equity Incentive Plan.
Indemnification
Indemnification of officers
Under Telix's Constitution, Telix has entered into agreements with each person who is, or has been, an officer of the
Company. This includes the Directors in office at the date of this report, the Company Secretary and other executive
officers, indemnifying them against any liability to any person other than Telix, or a related body corporate, that may
arise from their acting as officers of the Company, notwithstanding that they may have ceased to hold office. There is an
exception where the liability arises out of conduct involving a lack of good faith or is otherwise prohibited by law. During
and since the end of the financial year ended December 31, 2024, Telix has paid or agreed to pay the premiums for an
insurance policy to insure current and previous Directors and other executive officers against certain liabilities incurred in
that capacity. Due to the confidentiality obligations and undertakings set out in these agreements, no further details
regarding premiums paid, or the terms of the agreements, can be disclosed. No indemnity payment has been made
under any document referred to above during or since the financial year ended December 31, 2025.
Auditor independence and non-audit services
Telix may decide to employ its auditor on assignments additional to statutory audit duties where the auditor’s expertise
and experience with the Group are important.
Details of amounts paid or payable to Telix’s auditor, PricewaterhouseCoopers, for non-audit services provided during
the year are set out in note 37 of the Financial report. The Directors, in accordance with advice received from the Audit
and Risk Committee, are satisfied that the provision of non-audit services is compatible with the general standard of
independence for auditors imposed by the Australian Corporations Act 2001 (Cth) for the following reasons:
•the Audit and Risk Committee has reviewed, or if required pre-approved, all non-audit services to confirm they
do not affect the impartiality and objectivity of the auditor, and
•none of the services undermine the general principles relating to auditor independence as set out in APES 110
Code of Ethics for Professional Accountants, including reviewing or auditing the auditor’s work, acting in a
management or decision-making capacity for Telix, acting as an advocate for Telix, or jointly sharing the
economic risks and rewards.
A copy of the auditor’s independence declaration, as required under section 307C of the Australian Corporations Act
2001 (Cth), is included in this Annual Report as Exhibit 15.2.
Rounding
The Company is of a kind referred to in ASIC Legislative Instrument 2016/191, relating to the “rounding off” of amounts in
the Australian Directors’ report. Amounts in this report are rounded off in accordance with the instrument to the nearest
thousand dollars or, in certain cases, to the nearest dollar.
208
Corporate governance
Throughout 2025, Telix's governance arrangements were consistent with the 4th edition of the ASX Corporate
Governance Council’s Corporate Governance Principles and Recommendations. This year, Telix was unable to fully
comply with the ASX Corporate Governance Council's Corporate Governance Recommendation 1.5 due to new legal
requirements introduced in the U.S. Instead, Telix has provided a summary of its approach to belonging at Telix in the
2025 Corporate Governance Statement, which has been approved by the Board and available at ir.telixpharma.com/
governance/documents-charters. Telix is also subject to governance requirements related to our convertible bonds
listing on the SGX, and our Nasdaq listing and registration with the SEC outlined in Item 16G within this Annual Report.
Related party transactions with KMP
Remuneration: Remuneration to KMP is recorded in the tables above.
Loans: There were no loans between the Group and any KMP in the years ended December 31, 2025 and 2024.
Other transactions: Refer to note 36 of the Financial report for further details.
Other than those noted above, there were no related party transactions with any KMP in the year ended December 31,
2025.
The components of our Directors’ report are incorporated in various places within this Annual Report. Refer Contents
page for this Annual Report and table charting these components is included within Appendix 4E, which is included as
Exhibit 99.1 to this Annual Report.
Directors' Resolution
This report is approved in accordance with a resolution of the Directors.


Mark Nelson | Christian Behrenbruch |
Interim Chair | Managing Director and Group CEO |
February 20, 2026 | February 20, 2026 |
209
D.Employees
As of December 31, 2025, we had 1,184 employees based in 12 countries, as shown in the chart below.
Employees | |
United States | 886 |
Australia | 95 |
Belgium | 78 |
Canada | 48 |
Switzerland | 27 |
United Kingdom | 25 |
Japan | 10 |
France | 5 |
Spain | 6 |
The Netherlands | 2 |
Sweden | 1 |
New Zealand | 1 |
Total | 1,184 |
None of our employees are subject to a collective bargaining agreement. We consider our relationship with our
employees to be good.
E.Share Ownership
For information regarding the share ownership of our directors and executive officers, see “Item 7. Major Shareholders
and Related Party Transactions — A. Major Shareholders.”
F.Disclosure of a Registrant’s Action to Recover Erroneously Awarded Compensation
None.
210
ITEM 7. MAJOR SHAREHOLDERS AND RELATED PARTY TRANSACTIONS
A.Major Shareholders
The below table sets forth information with respect to the beneficial ownership of our ordinary shares as of
December 31, 2025, for:
•each person or group of affiliated persons known by us to beneficially own more than 5% of our ordinary shares;
•each of our executive officers;
•each of our directors; and
•all of our directors and executive officers as a group.
We have determined beneficial ownership in accordance with the rules and regulations of the SEC, and the information is
not necessarily indicative of beneficial ownership for any other purpose. Except as indicated by the footnotes below, we
believe, based on information furnished to us, that the persons and entities named in the table below have sole voting
and sole investment power with respect to all shares that they beneficially own.
Applicable percentage ownership is based on 338,777,049 ordinary shares outstanding as of December 31, 2025, which
includes 4,730,676 ADSs. As of December 31, 2025, we had 84 holders of record of our ordinary shares in the United
States, holding, in the aggregate 3,112,361 ordinary shares, or 0.92% of our outstanding ordinary shares, and 1 holder of
record of our ADSs in the United States, holding, in the aggregate, 4,330,676 ADSs, or 91.5% of our outstanding ADSs. In
computing the number of shares beneficially owned by a person or entity and the percentage ownership of such person
or entity, we deemed to be outstanding all shares subject to options held by the person or entity that are currently
exercisable, or exercisable within 60 days of December 31, 2025. However, except as described above, we did not deem
such shares outstanding for the purpose of computing the percentage ownership of any other person or entity. The
information contained in the following table is not necessarily indicative of beneficial ownership for any other purpose,
and the inclusion of any shares in the table does not constitute an admission of beneficial ownership of those shares.
Each of our shareholders is entitled to one vote per ordinary share. None of the holders of our ordinary have different
voting rights from other holders of our ordinary shares. We are not aware of any arrangement that may, at a subsequent
date, result in a change of control of our company. For further information regarding options to purchase ordinary shares
held by our directors and executive officers, see “Item 6. Directors, Senior Management and Employees — B.
Compensation.”
1 Based on information filed with the ASX by JPMorgan Chase & Co., and its affiliates on November 17, 2025. The address
for JPMorgan Chase & Co. is 270 Park Avenue, New York, NY, 10017.
2 Based on information filed with the ASX by State Street Corporation and subsidiaries on October 8, 2025. The address
for State Street Corporation is State Street Financial Center, 1 Lincoln Street, Boston, MA 02111.
3 Based on information filed with the ASX by Challenger Limited and its associated entities on September 25, 2025. The
address for Challenger Limited is Level 2, 5 Martin Place, Sydney NSW 2000, Australia.
4 Dr. Kluge, a former Non-Executive Director who retired in October 2024, is the Managing Director of Gnosis
Verwaltungsgesellschaft m.b.H. ("Gnosis") and has discretionary authority to vote and dispose of the ordinary shares
held by Gnosis. The address for Gnosis is Veilchenweg 38, 01326 Dresden, Germany.
5 Consists of (i) 21,329,006 ordinary shares beneficially owned (including 400,000 ADSs, each representing one of our
ordinary shares) and (ii) no ordinary shares issuable upon the exercise of options that are exercisable within 60 days of
December 31, 2025.
6 Consists of (i) 3,628,750 ordinary shares beneficially owned and (ii) no ordinary shares issuable upon the exercise of
options that are exercisable within 60 days of December 31, 2025.
7 Consists of (i) 106,550 ordinary shares beneficially owned and (ii) 52,070 ordinary shares issuable upon the exercise of
options that are exercisable within 60 days of December 31, 2025. Ms. Olson resigned as Chair and Non-Executive
Director effective February 3, 2026.
8 Consists of (i) 595,000 ordinary shares beneficially owned and (ii) no ordinary shares issuable upon the exercise of
options that are exercisable within 60 days of December 31, 2025.
9 Consists of (i) 3,719 ordinary shares beneficially owned and (ii) no ordinary shares issuable upon the exercise of options
that are exercisable within 60 days of December 31, 2025.
10 Consists of (i) 7,596 ordinary shares beneficially owned and (ii) no ordinary shares issuable upon the exercise of
options that are exercisable within 60 days of December 31, 2025.
11 Consists of (i) 25,558 ordinary shares beneficially owned and (ii) no ordinary shares issuable upon the exercise of
options that are exercisable within 60 days of December 31, 2025.
12 Consists of (i) 373,133 ordinary shares beneficially owned and (ii) no ordinary shares issuable upon the exercise of
options that are exercisable within 60 days of December 31, 2025.
211
Unless otherwise indicated, the address of each beneficial owner listed below is c/o Telix Pharmaceuticals Limited, 55
Flemington Road, North Melbourne, Victoria, 3051, Australia.
Name of Beneficial Owner | Number of Ordinary Shares Beneficially Owned | Percentage (%) |
5% or greater shareholders | ||
JPMorgan Chase & Co.1 | 17,663,335 | 5.21 |
State Street Corporation2 | 24,734,225 | 7.30 |
Challenger Limited3 | 18,096,417 | 5.34 |
Gnosis Verwaltungsgesellschaft m.b.H4 | 20,675,000 | 6.10 |
Directors and Executive officers | ||
Dr. Behrenbruch5 | 21,329,006 | 6.30 |
Mr. Nelson6 | 3,628,750 | 1.07 |
Ms. Olson7 | 158,620 | * |
Ms. Skinner8 | 595,000 | * |
Ms. McDonald9 | 3,719 | * |
Mr. Smith10 | 7,596 | * |
Dr. Patti11 | 25,558 | * |
Dr. Cade12 | 373,133 | * |
All directors and executive officers as a group (eight persons) | 26,121,382 | 7.71 |
*Less than one percent.
1 Total interest includes 400,000 ADSs.
2 H Kevin McCann retired as Non-Executive Director and Chairman of the Board on May 21, 2025.
3 Tiffany Olson resigned as Chair and Non-Executive Director effective February 3, 2026.
212
Australian Disclosure Requirements
Directors' interests in the securities of Telix
The relevant interests of each director in the share capital of Telix as at the date of this report are as follows:
Ordinary Shares | Options/PSARs | |
Dr. Behrenbruch1 | 21,329,006 | 526,497 |
Mr. McCann2 | - | - |
Mr. Nelson | 3,628,750 | - |
Ms. McDonald | 3,719 | - |
Ms. Olson3 | 106,550 | 52,070 |
Ms. Skinner | 595,000 | - |
B.Related Party Transactions
Since January 1, 2023, we have engaged in the following transactions with our directors, executive officers or holders of
more than 5% of our voting securities, or any member of the immediate family of, or person sharing the household with,
the foregoing persons, had or will have a direct or indirect material interest.
Director and Executive Officer Compensation
See “Item 6. Directors, Senior Management and Employees — B. Compensation” for information regarding compensation
of our executive officers and directors.
Master Services Agreement with ABX-CRO
ABX-CRO is a clinical research organization that specializes in radiopharmaceutical product development. We have
entered into a master services agreement with ABX-CRO for the provision of clinical and analytical services for its
programs. Dr. Andreas Kluge, a former non-executive director who retired from our board of directors on October 17,
2024, is the principal owner and Managing Director of ABX-CRO. In the year ended December 31, 2023, ABX-CRO was
engaged to perform close-out activities relating to the ZIRCON trial, and the total amount paid to ABX-CRO was
$835,315. During the year ended December 31, 2024, ABX-CRO continued to perform close out activities relating to the
ZIRCON trial, including delivery of dosimetry, PK evaluation, and the imaging report, and the total amount paid to ABX-
CRO was $513,809. The transactions with Andreas Kluge and ABX-CRO do not meet the criteria of a related party
transaction following Andreas Kluge's retirement as a non-executive director. Related party transactions were reviewed
on an ongoing basis by the Audit and Risk Committee in accordance with Australian law.
Consultancy Agreement with Dr. Andreas Kluge
Following Dr. Kluge’s retirement from our board of directors on October 17, 2024, Dr. Kluge was engaged by us on a
consultancy basis and will continue to provide the board of directors strategic advice alongside clinical input into key
development programs.
QDOSE Platform Partnership with ABX-CRO
In March 2024, we entered into an agreement to commercially partner the QDOSE dosimetry software platform with
ABX-CRO and its development partner, Quantinm AB. QDOSE is a software platform designed to enable reliable
estimation of patient-specific dosimetry for both therapeutic and diagnostic radiopharmaceuticals. We paid ABX-CRO
upfront cash consideration of €1.2 million, and agreed to pay a share of profits generated from QDOSE sales and a
referral fee on deals referred from or initiated by ABX-CRO for two years, expiring in 2026. During the year ended
December 31, 2024, no other payments were made to ABX-CRO pursuant to this agreement other than the initial upfront
cash consideration.
Indemnification Agreements
Our Constitution provides that, except to the extent prohibited by law including under the Australian Corporations Act
2001 (Cth), we must indemnify every person who is or has been a director, alternate director or executive officer of the
Company and such other officers or former officers of the Company or of its related bodies corporate as the board of
directors in each case determines against all losses, liabilities, costs, charges and expenses incurred by that person as a
director or officer.
1 Telix ASX disclosure July 22, 2025
213
We have entered into Deeds of Indemnity, Insurance and Access ("Indemnity Deeds") with Christian Behrenbruch, Marie
McDonald, Mark Nelson, Tiffany Olson, and Jann Skinner, and Deeds of Indemnity and Insurance with David Cade, Darren
Patti, and Darren Smith. Under the Indemnity Deeds, we have agreed to indemnify (to the maximum extent permitted
under Australian law and our Constitution, subject to certain specified exceptions) each director and executive officer
against all liabilities incurred in their capacity as our or our subsidiaries’ director or officer and any and all costs and
expenses relating to such a claim or to any notified event incurred by such director or executive officer, including costs
and expenses reasonably and necessarily incurred to mitigate any liability for such a claim or any claim which may arise
from such a notified event. The Indemnity Deeds provide that the indemnities are unlimited as to amount, continuous and
irrevocable.
Separately, we have obtained insurance for our directors and executive officers, as required by the Indemnity Deeds.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers or persons
controlling us pursuant to the foregoing provisions, we have been informed that in the opinion of the SEC such
indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable.
Related Party Transaction Policy
We comply with Australian law and the ASX Listing Rules regarding approval of transactions with related parties. Our
Audit and Risk Committee is responsible for reviewing and monitoring the propriety of related party transactions, as set
out in the Audit and Risk Committee Charter.
Effective upon the listing of our ADSs on Nasdaq, we amended our related party transaction policy to set forth our
procedures for the identification, review, consideration and approval or ratification of related party transactions to
comply with Nasdaq requirements. For purposes of our policy, a related party transaction is a transaction, arrangement
or similar contractual relationship, or any series of similar transactions, arrangements or relationships, in which we and
any related party are, were or will be participants and the amount involved in the transaction exceeds US$120,000, with
the exception of usual transactions concluded under normal conditions. A related party is any member of our board of
directors, our executive officers or any beneficial owner of more than 10% of any class of our ordinary shares, including
any of their immediate family members and any entity owned or controlled by such persons.
Under the policy, if a transaction has been identified as a related party transaction, including any transaction that was
not a related party transaction when originally consummated or any transaction that was not initially identified as a
related party transaction prior to consummation, our executive officers must present information regarding the related
party transaction to the chief financial officer and the transaction will be subject to review, consideration and approval by
the Audit and Risk Committee or, if required, the Board. The presentation must include a description of, among other
things, the material facts, the interests, direct and indirect, of the related party, the benefits to us of the transaction and
whether the transaction is on terms that are comparable to the terms available to or from, as the case may be, an
unrelated third-party or to or from employees generally. Under the policy, we will collect information that we deem
reasonably necessary from each member of our board of directors and executive officers to enable us to identify any
existing or potential related-person transactions and to effectuate the terms of the policy.
All of the transactions described above were entered into prior to the effective date of the amended written policy, but
our Board evaluated and approved all transactions that were considered to be related party transactions under Australian
law and the ASX Listing Rules at the time at which they were consummated.
C.Interests of Experts and Counsel
Not applicable.
ITEM 8. FINANCIAL INFORMATION
A.Consolidated Statements and Other Financial Information
See “Item 18. Financial Statements” for our consolidated financial statements filed as part of this Annual Report.
Legal Proceedings
As previously disclosed, Telix has received a subpoena from the SEC seeking various documents and information
primarily relating to the Company’s disclosures regarding the development of the Company’s prostate cancer therapeutic
candidates1. This matter remains a fact-finding request. Telix is cooperating with the subpoena including responding to
the SEC’s requests. The SEC has not asserted any charges against the Group or any of it's personnel, and no conclusions
have been reached, or that the SEC has a negative opinion of any person, entity or security. At this stage, the Group
cannot predict the duration, scope or outcome of this matter. The Group will continue to monitor the inquiry and update
its disclosures as appropriate.
On November 10, 2025, a putative securities class action complaint was filed in the United States District Court for the
Southern District of Indiana seeking, among other relief, class certification, designation of a lead plaintiff, damages and a
214
jury trial. On January 20, 2026, the Group was formally served the complaint on behalf of a purposed U.S. shareholder.
This case is in its earliest stages; no class has been certified and the court has not ruled on any dispositive motions. At
this stage, the Group cannot predict the duration, scope or outcome of this matter. The Group will continue to monitor
this matter and update its disclosures as appropriate.
Other than as set forth above, we are not currently a party to any material legal proceedings or investigations. From time
to time, we may become involved in other litigation or legal proceedings, particularly relevant to defending our IP rights
or in response to or relating to claims arising from the ordinary course of business.
Australian Disclosure Requirements
Dividends
No dividend was declared or paid, and Telix did not return capital to any of its shareholders, during the year ended
December 31, 2025.
Due to the stage of our company and the corporate objective of building and investing in our pipeline for the future, we
have not declared or paid any cash dividends on our ordinary shares and do not currently intend to do so for the
foreseeable future. We currently intend to invest our future earnings, if any, to fund our operations and pipeline
development activities and build the capabilities of our business to drive growth and value accretion. Future dividends, if
any, on our outstanding ordinary shares will be declared by and subject to the discretion of our Board, and subject to
applicable Australian law.
While we do not anticipate paying any cash dividends on our ordinary shares in the foreseeable future, if such a dividend
is declared, any dividend that we may declare will be paid to the holders of ADSs, subject to the terms of the deposit
agreement, to the same extent as holders of our ordinary shares, to the extent permitted by applicable law and
regulations, less the fees and expenses payable under the deposit agreement. Any dividend we declare will be
distributed by the depositary bank to the holders of the ADSs, subject to the terms of the deposit agreement. See “Item
12. Description of Securities Other Than Equity Securities — D. American Depositary Shares.”
B.Significant Changes
Except as otherwise disclosed in this Annual Report, no significant change has occurred since the date of the most
recent financial statements included in this Annual Report.
ITEM 9. THE OFFER AND LISTING
A.Offer and Listing Details
The principal trading market for our ordinary shares is the ASX in Australia, where our ordinary shares have been listed
since 2017. Our ordinary shares trade under the symbol “TLX.”
Since November 13, 2024, our ADSs have been listed on the Nasdaq Global Select Market, where they trade under the
symbol “TLX.” Each ADS represents one of our ordinary shares. JPMorgan Chase Bank, N.A., is the depositary for the
ADSs.
B.Plan of Distribution
Not applicable.
C.Markets
See “Item 9. The Offer and Listing — A. Offer and Listing Details.”
D.Selling Shareholders
Not applicable.
E.Dilution
Not applicable.
F.Expenses of the Issuer
Not applicable.
215
ITEM 10. ADDITIONAL INFORMATION
A.Share Capital
Not applicable.
B.Memorandum and Articles of Association
For Items 10.B.1 – 10.B.10, please refer to Item 10.B of our Registration Statement on Form 20-F, filed with the SEC on
October 29, 2024, which is incorporated into this Annual Report by reference.
C.Material Contracts
Please see “Item 4. Information on the Company — B. Business Overview — Collaboration and License Agreements” and
“Item 5. Operating and Financial Review and Prospects — B. Liquidity and Capital Resources” for a discussion of material
contracts. Except as otherwise disclosed in this Annual Report (including the exhibits) or the annual report for the year
ended December 31, 2024 that we filed with the SEC on February 24, 2025, we are not currently, and have not been in
the preceding two years, party to any material contract, other than contracts entered into in the ordinary course of
business.
D.Exchange Controls
Australia has largely abolished exchange controls on investment transactions. The Australian dollar is freely convertible
into U.S. dollars or other currencies. In addition, there are currently no specific rules or limitations regarding the export
from Australia of profits, dividends, capital or similar funds belonging to foreign investors, except that certain payments
to non-residents must be reported to the Australian Transaction Reports and Analysis Centre ("AUSTRAC"), which
monitors such transactions, and amounts on account of potential Australian tax liabilities may be required to be withheld
unless a relevant taxation treaty can be shown to apply and under such there are either exemptions or limitations on the
level of tax to be withheld.
E.Taxation
The following is a summary of material U.S. federal and Australian income tax considerations to U.S. Holders, as defined
below, of the acquisition, ownership and disposition of our ADSs and ordinary shares. This discussion is based on the
laws in force as of the date of this Annual Report, and is subject to changes in the relevant tax law, including changes
that could have retroactive effect. The following summary is not a comprehensive description of all U.S. federal or
Australian tax considerations that may be relevant to a decision to acquire or dispose of ADSs or ordinary shares and
does not take into account or discuss the tax laws of any country or other taxing jurisdiction other than the U.S. and
Australia. Holders are advised to consult their tax advisors concerning the overall tax consequences of the acquisition,
ownership and disposition of ADSs and ordinary shares in their particular circumstances. This discussion is not intended,
and should not be construed, as legal or professional tax advice.
Material U.S. Federal Income Tax Considerations
The following summary describes the material U.S. federal income tax consequences to a U.S. Holder (as defined below)
of the acquisition, ownership and disposition of the ADSs and ordinary shares as of the date hereof. This summary is
limited to U.S. Holders that hold the ADSs or ordinary shares as capital assets within the meaning of Section 1221 of the
Internal Revenue Code of 1986, as amended ("the Code").
This summary does not address the Medicare tax on net investment income, the effects of U.S. federal estate and gift
tax laws, alternative minimum taxes, or any state and local tax considerations. In addition, this section does not discuss
the tax consequences to any particular holder or any tax considerations that may apply to U.S. Holders subject to special
tax rules, such as:
•insurance companies;
•banks or other financial institutions;
•tax-exempt entities including pension plans, “individual retirement accounts” or “Roth IRAs”;
•regulated investment companies;
•real estate investment trusts;
•individuals who are former U.S. citizens or former long-term U.S. residents;
•brokers, dealers or traders in securities, commodities or currencies;
216
•traders that elect to use a mark-to-market method of accounting;
•except as specifically described below, persons holding the ADSs or ordinary shares through a partnership
(including an entity or arrangement treated as a partnership for U.S. federal income tax purposes) or S
corporation;
•persons that received ADSs or ordinary shares as compensation for the performance of services;
•persons that hold ADSs or ordinary shares as a position in a straddle or as part of a hedging, constructive sale,
conversion or other integrated transaction for U.S. federal income tax purposes;
•persons that have a functional currency other than the U.S. dollar;
•corporations that accumulate earnings to avoid U.S. federal income tax; or
•persons that own (directly, indirectly or constructively) 10% or more of our equity (by vote or value).
In this section, a “U.S. Holder” means a beneficial owner of our ADSs or ordinary shares that is, for U.S. federal income
tax purposes:
•an individual who is a citizen or resident of the U.S.;
•a corporation (or any other entity treated as a corporation for U.S. federal income tax purposes) created or
organized in or under the laws of the U.S. or any state thereof or the District of Columbia;
•an estate the income of which is subject to U.S. federal income taxation regardless of its source; or
•a trust (i) the administration of which is subject to the primary supervision of a court in the U.S. and for which
one or more U.S. persons has the authority to control all substantial decisions or (ii) that has an election in
effect under applicable U.S. Treasury regulations to be treated as a U.S. person for U.S. federal income tax
purposes.
We have not received, nor do we expect to seek, a ruling from the U.S. Internal Revenue Service ("IRS") regarding any
matter discussed herein. No assurance can be given that the IRS would not assert, or that a court would not sustain, a
position contrary to any of those set forth below. Each prospective investor should consult its own tax advisors with
respect to the U.S. federal, state and local and non-U.S. tax consequences of acquiring, owning and disposing of the
ADSs and ordinary shares.
If an entity or arrangement treated as a partnership for U.S. federal income tax purposes acquires, owns or disposes of
ADSs or ordinary shares, the U.S. federal income tax treatment of a partner in such partnership generally will depend on
the status of the partner and the activities of the partnership. Such a partner or partnership should consult its own tax
advisor as to the U.S. federal income tax consequences of acquiring, owning and disposing of the ADSs or ordinary
shares.
The discussion below is based upon the provisions of the Code, existing and proposed U.S. Treasury regulations,
published rulings and judicial decisions, all as of the date hereof. Such authorities may be replaced, revoked or modified,
possibly with retroactive effect, so as to result in U.S. federal income tax consequences different from those discussed
below. In addition, this summary is based, in part, upon representations made by the depositary to us and assumes that
the deposit agreement, and all other related agreements, will be performed in accordance with their terms.
YOU ARE URGED TO CONSULT YOUR OWN TAX ADVISOR WITH RESPECT TO THE U.S. FEDERAL, AS WELL AS STATE,
LOCAL AND NON-U.S. TAX CONSEQUENCES TO YOU OF ACQUIRING, OWNING AND DISPOSING OF ADSs OR
ORDINARY SHARES IN LIGHT OF YOUR PARTICULAR CIRCUMSTANCES, INCLUDING THE POSSIBLE EFFECTS OF
CHANGES IN U.S. FEDERAL AND OTHER TAX LAWS.
ADSs
Assuming that the representations contained in the deposit agreement are true and that the obligations in the deposit
agreement will be complied with in accordance with their terms, a U.S. Holder of ADSs generally will be treated for U.S.
federal income tax purposes as the owner of the underlying ordinary shares that are represented by such ADSs.
Accordingly, no gain or loss will be recognized for U.S. federal income tax purposes if a U.S. Holder exchanges ADSs for
the underlying ordinary shares represented by those ADSs.
Distributions
As described below in “– F. Dividends and Paying Agents,” we do not currently anticipate paying any distributions on the
ADSs or ordinary shares in the foreseeable future. However, to the extent there are any distributions made with respect
to the ADSs or ordinary shares, and subject to the PFIC rules discussed below, the gross amount of any such
distributions made out of our current or accumulated earnings and profits (as determined for U.S. federal income tax
purposes) will generally be taxable to a U.S. Holder as ordinary dividend income on the date such distribution is actually
217
or constructively received. Distributions in excess of our current and accumulated earnings and profits, as so
determined, will be treated first as a tax-free return of capital to the extent of the U.S. Holder’s adjusted tax basis in the
ADSs or ordinary shares, as applicable, and thereafter, as capital gain. However, because we do not intend to calculate
our earnings and profits under U.S. federal income tax principles, it is expected, and U.S. Holders should assume, that
any distribution will be reported as a dividend and will constitute ordinary dividend income to a U.S. Holder. Any
dividends will generally be treated as foreign-source and will not be eligible for the dividends-received deduction
generally allowed to corporate U.S. Holders.
Subject to the discussion under “—Passive Foreign Investment Company Considerations,” below, dividends paid to non-
corporate U.S. Holders may qualify as “qualified dividend income” eligible for the preferential rates of taxation applicable
to long-term capital gains if we are a “qualified foreign corporation” and certain other requirements (discussed below) are
met. We generally will be considered to be a qualified foreign corporation (i) if we are eligible for the benefits of the
Convention between the Government of the United States of America and the Government of Australia for the Avoidance
of Double Taxation and the Prevention of Fiscal Evasion with Respect to Taxes on Income, signed on August 6, 1982, as
amended and currently in force ("the U.S.-Australia Tax Treaty") or (ii) the ADSs or our ordinary shares are readily
tradable on an established securities market in the U.S. In addition, we believe that we qualify as a resident of Australia
for purposes of, and are eligible for the benefits of, the U.S.-Australia Tax Treaty, although there can be no assurance in
this regard. Therefore, subject to the discussion under “— Passive Foreign Investment Company Considerations,” below,
any dividends on the ADSs or our ordinary shares generally will be “qualified dividend income” in the hands of individual
U.S. Holders, provided that a holding period requirement (more than 60 days of ownership, without protection from the
risk of loss, during the 121-day period beginning 60 days before the ex-dividend date) and certain other requirements are
met.
Distributions paid in Australian dollars, including any Australian taxes withheld, will be included in a U.S. Holder’s gross
income in a U.S. dollar amount calculated by reference to the spot exchange rate in effect on the date of actual or
constructive receipt, regardless of whether the Australian dollars are converted into U.S. dollars at that time. A U.S.
Holder will have a tax basis in the Australian dollars equal to their U.S. dollar value on the date of receipt. As a result, if a
U.S. Holder converts the Australian dollars into U.S. dollars on the date of receipt, such U.S. Holder generally should not
be required to recognize any foreign exchange gain or loss. If Australian dollars so received are not converted into U.S.
dollars on the date of receipt, any gain or loss on a subsequent conversion or other disposition of the Australian dollars
generally will be treated as ordinary income or loss and generally will be income or loss from sources within the U.S. for
foreign tax credit limitation purposes.
Subject to certain limitations, a U.S. Holder may be able to claim as a credit against its U.S. federal income tax liability the
amount of any Australian tax withheld from any dividends at a rate not exceeding an applicable rate under the U.S.-
Australia Tax Treaty. Alternatively, a U.S. Holder may be able to deduct such Australian taxes from its U.S. federal
taxable income, provided that the U.S. Holder elects to deduct rather than credit all foreign income taxes paid or accrued
for the relevant taxable year. The rules governing U.S. foreign tax credits are complex and U.S. Treasury Regulations may
further restrict the availability of any such credit based on the nature of the withholding tax imposed by the foreign
jurisdiction. A notice from the IRS indicates that the IRS is considering proposing amendments to such foreign tax credit
regulations. Each U.S. Holder should consult its tax advisors regarding the foreign tax credit rules, including regarding
the availability of such credit or deductions.
Sale, Exchange or Other Disposition of ADSs or Ordinary Shares
A U.S. Holder generally will recognize gain or loss for U.S. federal income tax purposes upon the sale or other taxable
disposition of the ADSs or the ordinary shares in an amount equal to the difference between the U.S. dollar value of the
amount realized from such disposition and the U.S. Holder’s adjusted tax basis in those ADSs or ordinary shares,
determined in U.S. dollars. Subject to the discussion under “—Passive Foreign Investment Company Considerations”
below, any such gain or loss generally will be a capital gain or loss, and will be long-term capital gain or loss if the U.S.
Holder’s holding period for such ADSs or ordinary shares is more than one year at the time of such disposition. A U.S.
Holder’s adjusted tax basis in the ADSs or ordinary shares generally will be equal to the amount paid for such ADSs or
ordinary shares. Any long-term capital gain from the disposition of the ADSs or our ordinary shares by a non-corporate
U.S. Holder generally is eligible for a preferential rate of taxation. The deductibility of capital losses for U.S. federal
income tax purposes is subject to limitations. Any such gain or loss that a U.S. Holder recognizes generally will be treated
as U.S.-source gain or loss for foreign tax credit limitation purposes. U.S. Holders should consult their tax advisors
regarding the tax consequences if Australian taxes are imposed on or in connection with a sale, exchange or other
disposition of the ADSs or the ordinary shares and their ability to credit any Australian tax against their U.S. federal
income tax liability.
In the case of a U.S. Holder that is a cash basis taxpayer, any units of foreign currency received on a disposition of the
ADSs or our ordinary shares are translated into U.S. dollars at the spot exchange rate on the settlement date of the
disposition if the ADSs or ordinary shares disposed of are treated as traded on an established securities market. In such
case, no foreign currency exchange gain or loss will result for a cash basis taxpayer from currency fluctuations between
the trade date and the settlement date of such a disposition. An accrual basis taxpayer may elect the same treatment
required of cash basis taxpayers with respect to dispositions of the ADSs or our ordinary shares that are traded on an
established securities market, provided the election is applied consistently from year to year. Such election may not be
changed without the consent of the IRS. If an accrual basis taxpayer does not make such election, or if the ADSs or our
ordinary shares are not treated as traded on an established securities market, any units of foreign currency received on a
disposition of the ADSs or our ordinary shares are translated into U.S. dollars at the spot exchange rate on the trade date
218
of the disposition. In such case, the taxpayer may recognize exchange gain or loss based on currency fluctuations
between the trade date and the settlement date. Any foreign currency gain or loss a U.S. Holder recognizes will be U.S.-
source ordinary income or loss.
Passive Foreign Investment Company Considerations
If we are classified as a PFIC in any taxable year, certain adverse tax consequences could apply to U.S. Holders as a
result of that classification. We generally will be classified as a PFIC for any taxable year if (i) at least 75% of our gross
income for the taxable year consists of certain types of passive income, or (ii) at least 50% of our gross assets during the
taxable year, based on a quarterly average and generally determined by value, produce or are held for the production of
passive income. Passive income for this purpose generally includes, among other things, dividends, interest, rents,
royalties, gains from commodities and securities transactions and gains from the disposition of assets that produce or
are held for the production of passive income. Assets that produce or are held for the production of passive income
generally include cash, even if held as working capital or raised in a public offering, marketable securities and other
assets that may produce passive income. In determining whether we are a PFIC, we will be treated as owning our
proportionate share of the assets and earning our proportionate share of the income of each corporation in which we
own, directly or indirectly, at least a 25% interest (by value).
Based on the expected nature and amount of our estimated gross income, the anticipated nature and estimated average
value of our gross assets, the anticipated cash needs of our group’s operations and the nature and extent of the active
businesses conducted by our “25% or greater” owned subsidiaries, we do not expect that we will be classified as a PFIC
in the current taxable year or for the foreseeable future. However, the determination of our PFIC status for any taxable
year will not be determinable until after the end of the taxable year, and will depend on, among other things, the
composition of our income and assets (which could change significantly during the course of a taxable year) and the
market value of our assets for such taxable year, which may be, in part, based on the market price of the ADSs or
ordinary shares (which could be volatile). Accordingly, there can be no assurance that we will not be a PFIC for our
current or any future taxable year. U.S. Holders should consult their own tax advisors regarding our PFIC status.
If we are a PFIC for any taxable year during which a U.S. Holder holds ADSs or ordinary shares, absent certain elections
(including the mark-to-market election or qualified electing fund election described below), such U.S. Holder generally
will be subject to adverse rules (regardless of whether we continue to be classified as a PFIC) with respect to (i) any
“excess distribution” that we make to such U.S. Holder (generally, any distributions on the ADSs or ordinary shares in a
taxable year that are greater than 125% of the average annual distributions received by such U.S. Holder in the three
preceding taxable years or, if shorter, the U.S. Holder’s holding period) and (ii) any gain recognized from a sale or other
disposition (including a pledge) of such ADSs or ordinary shares. Under these special tax rules:
•the excess distribution or gain will be allocated ratably over the U.S. Holder’s holding period for the ADSs or
ordinary shares;
•the amount allocated to the current taxable year and any taxable year prior to the first taxable year in which we
were classified as a PFIC will be treated as ordinary income arising in the current taxable year (and would not
be subject to the interest charge discussed below); and
•the amount allocated to each other taxable year will be subject to income tax at the highest marginal tax rate in
effect for individuals or corporations, as applicable, for such year, and the interest charge generally applicable
to underpayments of tax will be imposed with respect to the resulting tax attributable to each such year.
In addition, non-corporate U.S. Holders will not be eligible for reduced rates of taxation applicable to “qualified dividend
income” on any dividends that we pay if we are a PFIC for either the taxable year in which the dividend is paid or the
preceding year.
If we are classified as a PFIC in any taxable year with respect to which a U.S. Holder owns ADSs or ordinary shares, we
generally will continue to be treated as a PFIC with respect to such U.S. Holder in all succeeding taxable years,
regardless of whether we continue to be classified as a PFIC under the tests described above, unless we cease to be
classified as a PFIC and such U.S. Holder makes a “deemed sale” election. If we cease to be classified as a PFIC and a
U.S. Holder makes the “deemed sale” election, such U.S. Holder will be deemed to have sold our ADSs or ordinary shares
at their fair market value on the last day of the last taxable year in which we were classified as a PFIC, and any gain
recognized from such deemed sale would be taxed under the PFIC excess distribution regime described above. After the
“deemed sale” election, a U.S. Holder’s ADSs or ordinary shares would not be treated as shares of a PFIC unless we
subsequently become a PFIC.
If we are a PFIC for any taxable year during which a U.S. Holder holds our ADSs or ordinary shares, and one of our non-
U.S. subsidiaries is also a PFIC (i.e., a lower-tier PFIC), such U.S. Holder would be treated as owning a proportionate
amount (by value) of the shares of the lower-tier PFIC and would be taxed under the PFIC excess distribution regime on
distributions by the lower-tier PFIC and on gain from the disposition of shares of the lower-tier PFIC even though such
U.S. Holder would not receive the proceeds of those distributions or dispositions.
If a U.S. Holder owns ADSs or our ordinary shares during any taxable year in which we are a PFIC, such U.S. Holder
generally will be required to file an IRS Form 8621 (Information Return by a Shareholder of a Passive Foreign Investment
219
Company or Qualified Electing Fund) with respect to us, generally with its U.S. federal income tax return for that year.
U.S. Holders should consult their tax advisors regarding any annual filing requirements.
If we are a PFIC, a U.S. Holder will not be subject to tax under the PFIC excess distribution regime on distributions or gain
recognized on our ADSs or ordinary shares if a valid “mark-to-market” election is made by the U.S. Holder for our ADSs
or ordinary shares, provided that the ADSs or ordinary shares held by such U.S. Holder are “marketable.”
If a U.S. Holder makes a mark-to-market election, it must include in gross income, as ordinary income, for each taxable
year that we are a PFIC an amount equal to the excess, if any, of the fair market value of the ADSs or ordinary shares
that are “marketable stock” at the close of the taxable year over such U.S. Holder’s adjusted tax basis in such ADSs or
ordinary shares. If a U.S. Holder makes such election, it may also claim a deduction as an ordinary loss in each such year
for the excess, if any, of such U.S. Holder’s adjusted tax basis in such ADSs or ordinary shares over their fair market
value at the end of the year, but only to the extent of the net amount previously included in income as a result of the
mark-to-market election. The U.S. Holder’s adjusted tax basis in the ADSs or ordinary shares with respect to which the
mark-to-market election applies would be adjusted to reflect amounts included in gross income or allowed as a
deduction because of such election. If a U.S. Holder makes an effective mark-to-market election, any gain recognized
upon the sale or other disposition of the ADSs or ordinary shares in a year that we are a PFIC will be treated as ordinary
income and any loss will be treated first as ordinary loss (to the extent of any net mark-to-market gains for prior years)
and thereafter as capital loss. However, a mark-to-market election will generally not be available with respect to a lower-
tier PFIC unless the shares of such lower-tier PFIC are themselves treated as “marketable stock.”
If a U.S. Holder makes a mark-to-market election, it will be effective for the taxable year for which the election is made
and all subsequent taxable years unless the ADSs or ordinary shares are no longer regularly traded on a qualified
exchange or the IRS consents to the revocation of the election. U.S. Holders are urged to consult their tax advisors about
the availability of the mark-to-market election.
Alternatively, in certain cases, a U.S. Holder may be able to avoid the interest charge and the other adverse PFIC tax
consequences described above by electing to treat the PFIC as a “qualified electing fund,” or QEF, under Section 1295 of
the Code. If a U.S. Holder makes a valid and timely QEF election and we provide certain required information to such U.S.
Holder, then for each taxable year to which such an election applies, the U.S. Holder will be subject to U.S. federal
income tax on its pro rata share of our net capital gain and ordinary earnings, regardless of whether such amounts are
actually distributed to the U.S. Holder in that year or any later year. However, we do not anticipate that this election will
be available to U.S. Holders because we do not expect to provide U.S. Holders with the information that would be
necessary to make a valid QEF election.
Backup Withholding Tax and Information Reporting Requirements
U.S. Holders generally will be subject to information reporting requirements with respect to distributions paid on the
ADSs or our ordinary shares, and on the proceeds from the sale, exchange or other disposition of the ADSs or our
ordinary shares that are paid within the U.S. or through U.S.-related financial intermediaries, unless the U.S. Holder is an
“exempt recipient.” In addition, U.S. Holders may be subject to backup withholding on such payments, unless the U.S.
Holder provides a correct taxpayer identification number and a duly executed IRS Form W-9 or otherwise establishes an
exemption. Backup withholding is not an additional tax, and the amount of any backup withholding will be allowed as a
credit against a U.S. Holder’s U.S. federal income tax liability and may entitle such holder to a refund, provided that the
required information is timely furnished to the IRS.
Certain U.S. Holders are required to report information relating to an interest in the ADSs and our ordinary shares, subject
to certain exceptions (including an exception for ADSs and ordinary shares held in accounts maintained by U.S. financial
institutions) by filing IRS Form 8938 (Statement of Specified Foreign Financial Assets) with their U.S. federal income tax
return. Substantial penalties may be imposed upon a U.S. Holder that fails to comply. U.S. Holders should consult their
tax advisors regarding their information reporting obligations, if any, with respect to their ownership and disposition of
the ADSs or our ordinary shares.
THE DISCUSSION ABOVE IS A SUMMARY OF THE MATERIAL U.S. FEDERAL INCOME TAX CONSEQUENCES OF AN
INVESTMENT IN THE ADSs AND ORDINARY SHARES AND IS BASED UPON LAWS AND RELEVANT INTERPRETATIONS
THEREOF IN EFFECT AS OF THE DATE OF THIS ANNUAL REPORT, ALL OF WHICH ARE SUBJECT TO CHANGE OR
DIFFERING INTERPRETATION, POSSIBLY WITH RETROACTIVE EFFECT. EACH PROSPECTIVE INVESTOR SHOULD
CONSULT ITS TAX ADVISOR ABOUT THE TAX CONSEQUENCES TO IT OF AN INVESTMENT IN THE ADSs AND
ORDINARY SHARES IN LIGHT OF THE INVESTOR’S OWN CIRCUMSTANCES.
Material Australian Tax Considerations
In this section, we discuss the material Australian income tax, stamp duty and goods and services tax considerations
related to the acquisition, ownership and disposal by the absolute beneficial owners of the ADSs or ordinary shares. It is
based upon existing Australian tax law as of the date of this Annual Report, which is subject to change, possibly
retrospectively. This discussion does not address all aspects of Australian tax law which may be important to particular
investors in light of their individual investment circumstances, such as ADSs or shares held by investors subject to
special tax rules (for example, financial institutions, insurance companies or tax-exempt organizations). In addition, this
summary does not discuss any non-Australian or state tax considerations, other than stamp duty.
220
Prospective investors are urged to consult their tax advisors regarding the Australian and non-Australian income and
other tax considerations of the acquisition, ownership and disposition of the ADSs or shares. This summary is based
upon the premise that the holder is not an Australian tax resident and is not carrying on business in Australia through a
permanent establishment (referred to as a “Non-Australian Holder” in this summary).
Nature of ADSs for Australian Taxation Purposes
Non-Australian Holders of ADSs should obtain specialist Australian tax advice regarding their rights and obligations under
the deposit agreement with the depositary, including whether the deposit arrangement constitutes a ‘bare trust’ for
Australian taxation purposes. Specialist Australian tax advice should also be obtained before a Non-Australian Holder
surrenders ADSs to the depository for cancellation to receive the ordinary shares underlying those ADSs. Apart from
certain aspects of the Australian tax legislation (for example, the Australian capital gains tax and withholding tax
provisions, which are discussed below), there is no express legislative basis for disregarding “bare trusts” for Australian
tax purposes generally. This summary proceeds on the assumption that the deposit arrangement constitutes a bare trust
such that a Holder of an ADS is absolutely entitled (as against the depository) to the underlying share and presently
entitled to dividends paid on the underlying shares.
Holders of ADSs can be treated as the owners of the underlying ordinary shares for Australian capital gains tax purposes
provided that they are ‘absolutely entitled’ to those shares. Dividends paid on the underlying ordinary shares will also be
treated as dividends derived by the holders of ADSs as the persons presently entitled to those dividends.
Taxation of Dividends
Australia operates a dividend imputation system under which dividends may be declared to be “franked” to the extent
they are paid out of company profits that have been subject to income tax. Fully franked dividends are not subject to
dividend withholding tax. To the extent that they are unfranked, dividends payable to Non-Australian Holders will be
subject to dividend withholding tax except to the extent they are declared to be conduit foreign income ("CFI"). Dividend
withholding tax will be imposed at 30%, unless a shareholder is a resident of a country with which Australia has a double
taxation treaty and qualifies for the benefits of the treaty. Under the provisions of the current Double Taxation
Convention between Australia and the U.S., the Australian tax withheld on unfranked dividends that are not declared to
be CFI to which a resident of the U.S. is beneficially entitled is limited to 15% where that resident is a qualified person for
the purposes of the Double Taxation Convention.
Under the Double Taxation Convention between Australia and the U.S., if a company that is a resident of the U.S. and
qualifies for the benefits of the Convention directly owns a 10% or greater interest in us, the Australian tax withheld on
unfranked dividends that are not declared to be CFI paid by us to which the company is beneficially entitled is limited to
5%.
Character of ADSs for Australian Taxation Purposes
The Australian tax treatment of a sale or disposal of the ADSs will depend on whether they are held on revenue or capital
account. ADSs may be held on revenue rather than capital account, for example, where they are held by share traders or
where the shares are acquired for the purposes of sale by the holder at a profit. Non-Australian Holders of ADSs should
obtain specialist Australian tax advice regarding the characterization of any gain or loss on a sale or disposal of the ADSs
as revenue or capital in nature.
Tax on Sales or other Dispositions of Shares—Capital Gains Tax
A Non-Australian Holder who is treated as the owner of the underlying shares on the basis that they are absolutely
entitled to those shares will not be subject to Australian capital gains tax on the gain made on a sale or other disposal of
ordinary shares unless the shares are “taxable Australian property.” The shares will be “taxable Australian property”
under current law where:
•the Non-Australian Holder, together with associates, holds 10% or more of our issued capital, at the time of
disposal or for a 12-month period during the two years prior to disposal; and
•more than 50% of our assets held directly or indirectly, determined by reference to market value, consist of
Australian real property (which includes land and leasehold interests) or Australian mining, quarrying or
prospecting rights at the time of disposal.
However, the Australian government announced that the capital gains tax rules for non-residents will be clarified and
broadened with effect from July 1, 2025 so that they apply to assets with ‘a close economic connection to Australian
land’ (in addition to ‘real property’), and to apply the 50% value test throughout a 12 month period prior to disposal rather
than at the time of disposal. Non-Australian Holders should monitor developments in this regard.
Australian capital gains tax applies to net capital gains at a taxpayer’s marginal tax rates. Net capital gains are calculated
after reduction for capital losses, which may only be offset against capital gains. The 50% capital gains tax discount is
not available to Non-Australian Holders on gains from assets acquired or accrued after May 8, 2012 where they were
non-Australian residents during the entire holding period. Companies are not entitled to a capital gains tax discount.
221
Broadly, where there is a disposal of certain taxable Australian property, the purchaser will be required to withhold and
remit to the Australian Taxation Office ("ATO") 12.5% of the proceeds from the sale. On December 13, 2023, the
Australian government announced that the withholding rate will be increased from 12.5% to 15% of the proceeds of sale
for disposals occurring from January 1, 2025. While draft legislation has been released, this announced increase is yet to
be legislated and may be subject to change. A transaction is excluded from the withholding requirements in certain
circumstances, including where the transaction is an on-market transaction conducted on an approved stock exchange,
a securities lending transaction, or the transaction is conducted using a broker operated crossing system. There may
also be an exception to the requirement to withhold where a Non-Australian Holder provides a declaration that their
ordinary shares are not ‘indirect Australian real property interests,’ although the Australian government is currently
running a consultation process to consider whether the Australian Taxation Office should be notified in advance of such a
declaration being made for transactions with a value in excess of A$20 million. The Non-Australian Holder may be
entitled to receive a tax credit for the tax withheld by the purchaser which they may claim in their Australian income tax
return.
Tax on Sales or other Dispositions of ADSs or Shares—Revenue Account
Non-Australian Holders who hold their ADSs on revenue account may have the gains made on the sale or other disposal
of the ADSs included in their assessable income under the ordinary income provisions of the income tax law, if the gains
are sourced in Australia. There are no express provisions which treat holders of ADSs as the owners of the underlying
shares where there is a bare trust.
Non-Australian Holders assessable under these ordinary income provisions in respect of gains made on ADSs held on
revenue account would be assessed for such gains at the Australian tax rates for non-Australian residents, which start at
a marginal rate of 30% for individuals, and would be required to file an Australian tax return. Relief from Australian income
tax may be available to a Non-Australian Holder who is resident of a country with which Australia has a double taxation
treaty, qualifies for the benefits of the treaty and does not, for example, derive the gain in carrying on business through a
permanent establishment in Australia. To the extent an amount would be included in a Non-Australian Holder’s
assessable income under both the capital gains tax provisions and the ordinary income provisions, the capital gain
amount may be reduced, so that the holder may not be subject to double Australian tax on any part of the gain.
The statements under “— Tax on Sales or Other Dispositions of Shares—Capital Gains Tax” regarding a purchaser being
required to withhold 12.5% tax (proposed to increase to 15% from January 1, 2025) on the acquisition of certain taxable
Australian property are also relevant where the disposal of the ADSs by a Non-Australian Holder is likely to generate
gains on revenue account, rather than a capital gain.
The same consequences apply for Non-Australian Holders who hold ordinary shares on revenue account.
Dual Residency
If a holder of ADSs is a resident of both Australia and the U.S. under those countries’ domestic taxation laws, that holder
may be subject to tax as an Australian resident. If, however, the holder is an individual and is determined to be a U.S.
resident for the purposes of the Double Taxation Convention between the U.S. and Australia, the Australian tax would be
subject to limitation by the Double Taxation Convention. Holders should obtain specialist taxation advice in these
circumstances.
Stamp Duty
No Australian stamp duty is payable by Australian residents or non-Australian residents on the issue, transfer and/or
surrender of the ADSs or ordinary shares while we continue to satisfy the requirements of a listed company for the
purposes of Australian duties legislation, provided that the securities issued, transferred and/or surrendered do not
represent 90% or more of our issued shares.
Australian Death Duty
Australia does not have estate or death duties. As a general rule, no capital gains tax liability is realized upon the
inheritance of a deceased person’s shares. The disposal of inherited shares by beneficiaries may, however, give rise to a
capital gains tax liability if the gain falls within the scope of Australia’s jurisdiction to tax.
Goods and Services Tax
No Australian goods and services tax will be payable on the supply of the ADSs or ordinary shares.
THE DISCUSSION ABOVE IS A SUMMARY OF THE AUSTRALIAN TAX CONSEQUENCES OF AN INVESTMENT IN OUR
ORDINARY SHARES OR ADSs AND IS BASED UPON LAWS AND RELEVANT INTERPRETATIONS THEREOF IN EFFECT AS
OF THE DATE OF THIS ANNUAL REPORT, ALL OF WHICH ARE SUBJECT TO CHANGE, POSSIBLY WITH RETROACTIVE
EFFECT. EACH PROSPECTIVE INVESTOR IS URGED TO CONSULT ITS OWN TAX ADVISOR ABOUT THE TAX
CONSEQUENCES TO IT OF AN INVESTMENT IN OUR ORDINARY SHARES OR ADSs IN LIGHT OF THE INVESTOR’S OWN
CIRCUMSTANCES.
222
F.Dividends and Paying Agents
Not applicable.
G.Statement by Experts
Not applicable.
H.Documents on Display
We are subject to the reporting requirements of the Exchange Act, as applicable to “foreign private issuers” as defined in
Rule 3b-4 under the Exchange Act. See “Item 6. Directors, Senior Management and Employees — C. Board Practices —
Foreign Private Issuer Exemption” for information on reporting exemptions applicable to us as a foreign private issuer. In
addition, we are not required under the Exchange Act to file annual, quarterly and current reports and financial
statements with the SEC as frequently or as promptly as U.S. domestic companies whose securities are registered under
the Exchange Act. However, we file with the SEC an annual report on Form 20-F containing financial statements audited
by an independent registered public accounting firm, and submit reports to the SEC on Form 6-K to disclose other
material information, as required. We also submit reports to the SEC on Form 6-K containing unaudited financial
information for the first six months of each fiscal year.
The SEC maintains a website at www.sec.gov from which our filings may be accessed.
In addition, since our ordinary shares are traded on the ASX, we have filed periodic corporate reports, including annual
and semi-annual reports with, and furnish information to, the ASX, as required under the ASX Listing Rules and the
Australian Corporations Act 2001 (Cth). Copies of our filings with the ASX can be retrieved electronically at
www.asx.com.au under our symbol “TLX”. We also maintain a website at www.telixpharma.com.
I.Subsidiary Information
For information about our subsidiaries, see “Item 4. Information on the Company — C. Organizational Structure.”
ITEM 11. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT
MARKET RISK
We are exposed to market risks in the ordinary course of our business. Market risk represents the risk of loss that may
impact our financial position due to adverse changes in financial market prices and rates. Our market risk exposure is
primarily attributable to foreign currency exchange rate risk.
Interest Rate Risk
As of December 31, 2025, we had cash and cash equivalents of $141.9 million. We have limited exposure to interest rate
risk. Our cash and cash equivalents are not locked into long-term deposits at fixed rates so as to mitigate the risk of
earning interest below the current floating rate.
Our exposure to market interest rates relates primarily to short-term deposits. The roll-over loan facility totaling $9.5
million (translated from Euros based on the exchange currency rate as of December 31, 2025) carries an interest rate
that is calculated using the eurozone interbank interest rate as of each interest determination date. However, all of our
borrowings that have been drawn down as of December 31, 2025 bear a fixed interest rate. Therefore, we are not
exposed to any significant interest rate risk under these borrowings. An immediate 10% change in current market interest
rates would not have a material impact on our borrowings, financial position or results of operations.
We do not believe that inflation has had a material effect on our business, financial condition, or results of operations.
Nonetheless, if our costs were to become subject to significant inflationary pressures, we may not be able to fully offset
such higher costs. Our inability or failure to do so could harm our business, results of operations, or financial condition.
Foreign Currency Exchange Rate Risk
Foreign currency risk is the risk of fluctuation in fair value or future cash flows of a financial instrument as a result of
changes in foreign exchange rates. We operate internationally and are exposed to foreign exchange risk, primarily
related to the Australian dollar and Euro.
Our treasury risk management policy is to settle all U.S. dollar denominated expenditures with U.S. dollar denominated
receipts from sales of Commercial Products in the U.S. We also manage currency risk by making decisions as to the
levels of cash to hold in each currency by assessing future activities which will likely be incurred in those currencies. Any
remaining foreign currency exposure has therefore not been hedged.
223
We have both foreign currency receivables and payables, predominantly denominated in Australian dollar and Euro. We
had a surplus of foreign currency receivables and financial assets over payables of $67.4 million as of December 31,
2025.
Our exposure to the risk of changes in foreign exchange rates also relates to the net investments in foreign subsidiaries,
which predominantly include denominations in Euro and Australian dollar, however given the level of current investments
in non US dollar denominated subsidiaries, the impact is limited
As of December 31, 2025, we held 39.7% of our cash in U.S. dollars, 52.8% in Australian dollars, 5.5% in Euros, 0.3% in
British pounds, 0.5% in Canadian dollars and 1.0% in Swiss Francs. The following table sets forth the balances of our cash
and cash equivalents, trade receivables and financial assets as of December 31, 2025 that give rise to currency risk
exposure, as presented in US$:
USD | AUD | EUR | CHF | JPY | GBP | CAD | |
U.S. Dollars | Australian Dollars | Euros | Swiss Francs | Japanese Yen | British Pounds | Canadian Dollars | |
US$'000 | US$'000 | US$'000 | US$'000 | US$'000 | US$'000 | US$'000 | |
Cash and cash equivalents | 56,371 | 74,891 | 7,850 | 1,445 | 134 | 413 | 762 |
Trade receivables | 126,056 | 540 | 2,606 | - | - | - | - |
Financial assets | - | 33,749 | 3,345 | - | - | - | - |
We are primarily exposed to foreign exchange risk inherent in Australian dollar-denominated cash and cash equivalents,
trade receivables, trade payables and financial assets and in Euro-denominated cash and cash equivalents, trade
payables and contingent consideration liability. We also have exposure to exchange rate risk from the Euro attributable
to our Euro-denominated loans from BNP Paribas and IMBC Group. For the year ended December 31, 2025, an increase
or decrease of the US dollar to Australian dollar exchange rate by +10% would decrease our profit after tax by $nil or
decrease our profit after tax by $nil, respectively, and an increase or decrease of the US$ to Euro exchange rate by +10%
would decrease our profit after tax by US$0.2 million or increase our profit after tax by US$2.0 million, respectively. For
the year ended December 31, 2024, an increase or decrease of the U.S. dollar to Australian dollar exchange rate by +10%
would decrease our profit after tax by $nil or increase our profit after tax by $nil, respectively, and an increase or
decrease of the US dollar to Euro exchange rate by +10% would increase our profit after tax by US$1.2 million or
decrease our profit after tax by US$1.5 million, respectively. For more information on our currency risk exposure and
sensitivity analysis, see Note 33.3 to our audited consolidated financial statements included elsewhere in this Annual
Report.
Liquidity Risk
We are exposed to liquidity and funding risk from operations and from external borrowings, where the risk is that we may
not be able to refinance debt obligations or meet other cash outflow obligations when required. Vigilant liquidity risk
management requires that we maintain sufficient liquid assets (mainly cash and cash equivalents). We manage liquidity
risk by maintaining adequate cash reserves by continuously monitoring actual and forecast cash flows and matching the
maturity profiles of financial assets and liabilities.
Credit Risk
Credit risk refers to the risk that a counterparty will default on its contractual obligations resulting in financial loss to us.
Credit risk arises from cash and cash equivalents and credit exposures to customers, including outstanding receivables.
Credit risk is managed on a group basis. If customers are independently rated, these ratings are used. Otherwise, if there
is no independent rating, we assess the credit quality of the customer, taking into account its financial position, past
experience and other factors. Individual risk limits are set based on internal or external ratings. The compliance with
credit limits by customers is regularly monitored. We obtain guarantees where appropriate to mitigate credit risk.
We apply the IFRS 9 simplified approach to measuring expected credit losses which uses a lifetime expected loss
allowance for all trade receivables.
To measure the expected credit losses, trade receivables have been grouped based on shared credit risk characteristics
and the days past due. The expected loss rates are based on historical payment profiles of sales and the corresponding
historical credit losses experienced. The historical loss rates are adjusted to reflect current and forward-looking
information on macroeconomic factors affecting the ability of the customers to settle the receivables.
Trade receivables are written off where there is no reasonable expectation of recovery. Indicators that there is no
reasonable expectation of recovery include, amongst others, the failure of a debtor to engage in a repayment plan with
us, and the failure to make contractual payments for a period of greater than 120 days past due.
224
Impairment losses on trade receivables are presented within sales and marketing costs within profit or loss. Subsequent
recoveries of amounts previously written off are credited against the same line item. The expected credit losses were
$2.4 million and $0.1 million as of December 31, 2025 and 2024 respectively.
ITEM 12.DESCRIPTION OF SECURITIES OTHER THAN EQUITY SECURITIES
A.Debt Securities
Not applicable.
B.Warrants and Rights
Not applicable.
C.Other Securities
Not applicable.
D.American Depositary Shares
Fees and Expenses
Our ADS depositary, JPMorgan Chase Bank, N.A. ("JPMorgan") may charge each person to whom ADSs are issued,
including, without limitation, issuances against deposits of ordinary shares, issuances in respect of share distributions,
rights and other distributions, issuances pursuant to a stock dividend or stock split declared by us or issuances pursuant
to a merger, exchange of securities or any other transaction or event affecting the ADSs or deposited securities, and
each person surrendering ADSs for withdrawal of deposited securities or whose ADSs are cancelled or reduced for any
other reason, a fee of up to US$5.00 for each 100 ADSs (or any portion thereof) issued, delivered, reduced, cancelled or
surrendered, or upon which a share distribution or elective distribution is made or offered, as the case may be.
The depositary may sell (by public or private sale) sufficient securities and property received in respect of a share
distribution, rights and/or other distribution prior to such deposit to pay such charge.
The following additional fees, charges and expenses shall also be incurred by the ADR holders, the beneficial owners, by
any party depositing or withdrawing ordinary shares or by any party surrendering ADSs and/or to whom ADSs are issued
(including, without limitation, issuance pursuant to a stock dividend or stock split declared by us or an exchange of stock
regarding the ADSs or the deposited securities or a distribution of ADSs), whichever is applicable:
•a fee of up to US$0.05 per ADS held for any cash distribution made, or for any elective cash/stock dividend offered,
pursuant to the deposit agreement;
•an aggregate fee of up to US$0.05 per ADS per calendar year (or portion thereof) for services performed by the
depositary in administering the ADRs (which fee may be charged on a periodic basis during each calendar year and
shall be assessed against holders of ADRs as of the record date or record dates set by the depositary during each
calendar year and shall be payable in the manner described in the next succeeding provision);
•an amount for the reimbursement of such fees, charges and expenses as are incurred by the depositary and/or any
of its agents (including, without limitation, the custodian, as well as charges and expenses incurred on behalf of
ADR holders in connection with compliance with foreign exchange control regulations or any law, rule or regulation
relating to foreign investment) in connection with the servicing of the ordinary shares or other deposited securities,
the sale of securities (including, without limitation, deposited securities), the delivery of deposited securities or
otherwise in connection with the depositary’s or its custodian’s compliance with applicable law, rule or regulation
(which fees and charges shall be assessed on a proportionate basis against ADR holders as of the record date or
dates set by the depositary and shall be payable at the sole discretion of the depositary by billing such ADR holders
or by deducting such charge from one or more cash dividends or other cash distributions);
•a fee of up to US$0.05 per ADS held for the direct or indirect distribution of securities (other than ADSs or rights to
purchase additional ADSs) or the net cash proceeds from the public or private sale of such securities, regardless of
whether any such distribution and/or sale is made by, for, or received from, or (in each case) on behalf of, the
depositary, us and/or any third-party (which fee may be assessed against ADR holders as of a record date set by
the depositary);
•stock transfer or other taxes and other governmental charges;
•a transaction fee per cancellation request (including any cancellation request made through SWIFT, facsimile
transmission or any other method of communication) as disclosed on the “Disclosures” page (or successor page) of
www.adr.com (as updated by the depositary from time to time, “ADR.com”) and any applicable delivery expenses
(which are payable by such persons or ADR holders);
225
•transfer or registration fees for the registration of transfer of deposited securities on any applicable register in
connection with the deposit or withdrawal of deposited securities; and
•fees of any division, branch or affiliate of the depositary utilized by the depositary to direct, manage and/or execute
any public and/or private sale of securities under the deposit agreement.
To facilitate the administration of various depositary receipt transactions, including disbursement of dividends or other
cash distributions and other corporate actions, the depositary may engage the foreign exchange desk within the banking
division of JPMorgan ("the Bank") and/or its affiliates in order to enter into spot foreign exchange transactions to convert
foreign currency into U.S. dollars. For certain currencies, foreign exchange transactions are entered into with the Bank or
an affiliate, as the case may be, acting in a principal capacity. For other currencies, foreign exchange transactions are
routed directly to and managed by an unaffiliated local custodian (or other third-party local liquidity provider), and
neither the Bank nor any of its affiliates is a party to such foreign exchange transactions.
The foreign exchange rate applied to a foreign exchange transaction will be either (i) a published benchmark rate, or (ii) a
rate determined by a third-party local liquidity provider, in each case plus or minus a spread, as applicable. The
depositary will disclose which foreign exchange rate and spread, if any, apply to such currency on the “Disclosures” page
(or successor page) of ADR.com. Such applicable foreign exchange rate and spread may (and neither the depositary, the
Bank nor any of their affiliates is under any obligation to ensure that such rate does not) differ from rates and spreads at
which comparable transactions are entered into with other customers or the range of foreign exchange rates and
spreads at which the Bank or any of its affiliates enters into foreign exchange transactions in the relevant currency pair
on the date of the foreign exchange transaction. Additionally, the timing of execution of a foreign exchange transaction
varies according to local market dynamics, which may include regulatory requirements, market hours and liquidity in the
foreign exchange market or other factors. Furthermore, the Bank and its affiliates may manage the associated risks of
their position in the market in a manner they deem appropriate without regard to the impact of such activities on the
depositary, us, ADR holders or beneficial owners. The spread applied does not reflect any gains or losses that may be
earned or incurred by the Bank and its affiliates as a result of risk management or other hedging related activity.
Notwithstanding the foregoing, to the extent we provide U.S. dollars to the depositary, neither the Bank nor any of its
affiliates will execute a foreign exchange transaction as set forth herein. In such case, the depositary will distribute the
U.S. dollars received from us.
Further details relating to the applicable foreign exchange rate, the applicable spread and the execution of foreign
exchange transactions will be provided by the depositary on ADR.com. Each holder and beneficial owner by holding or
owning an ADR or ADS or an interest therein, and we, each acknowledge and agree that the terms applicable to foreign
exchange transactions disclosed from time to time on ADR.com will apply to any foreign exchange transaction executed
pursuant to the deposit agreement.
We will pay all other fees, charges and expenses of the depositary and any agent of the depositary (except the
custodian) pursuant to agreements from time to time between us and the depositary.
The right of the depositary to charge and receive payment of fees, charges and expenses survives the termination of the
deposit agreement, and shall extend for those fees, charges and expenses incurred prior to the effectiveness of any
resignation or removal of the depositary.
The fees and charges described above may be amended from time to time by agreement between us and the
depositary.
Depositary Reimbursements
The depositary anticipates reimbursing us for certain expenses incurred by us that are related to the establishment and
maintenance of the ADR program upon such terms and conditions as we and the depositary may agree from time to time.
The depositary may make available to us a set amount or a portion of the depositary fees charged in respect of the ADR
program or otherwise upon such terms and conditions as we and the depositary may agree from time to time. The
depositary may also agree to reduce or waive certain fees that would normally be charged on ADSs issued to or at the
direction of, or otherwise held by, us and/or certain holders and beneficial owners and holders and beneficial owners of
shares of ours. The depositary collects its fees for issuance and cancellation of ADSs directly from investors depositing
shares or surrendering ADSs for the purpose of withdrawal or from intermediaries acting for them. The depositary
collects fees for making distributions to investors by deducting those fees from the amounts distributed or by selling a
portion of distributable property to pay the fees. The depositary may collect its annual fee for depositary services by
deduction from cash distributions, or by directly billing investors, or by charging the book-entry system accounts of
participants acting for them. The depositary will generally set off the amounts owing from distributions made to holders
of ADSs. If, however, no distribution exists and payment owing is not timely received by the depositary, the depositary
may refuse to provide any further services to ADR holders that have not paid those fees and expenses owing until such
fees and expenses have been paid. At the discretion of the depositary, all fees and charges owing under the deposit
agreement are due in advance and/or when declared owing by the depositary.
226
Payment of Taxes
ADR holders and/or beneficial owners must pay any tax or other governmental charge payable by the custodian or the
depositary on any ADS or ADR, deposited security or distribution. If any taxes or other governmental charges (including
any penalties and/or interest) shall become payable by or on behalf of the custodian or the depositary with respect to
any ADR, any deposited securities represented by the ADSs evidenced thereby or any distribution thereon such tax or
other governmental charge shall be paid by the ADR holder thereof to the depositary and by holding or owning, or having
held or owned, an ADR or any ADSs evidenced thereby, the ADR holder and all beneficial owners thereof, and all prior
ADR holders and beneficial owners thereof, jointly and severally, agree to indemnify, defend and save harmless each of
the depositary and its agents in respect of such tax or other governmental charge. Notwithstanding the depositary’s right
to seek payment from current or former ADR holders and beneficial owners, each ADR holder and beneficial owner, and
each prior ADR holder and beneficial owner, by holding or owning, or having held or owned, an ADR or an interest in
ADSs acknowledges and agrees that the depositary has no obligation to seek payment of amounts owing from any
current or prior beneficial owner. If an ADR holder owes any tax or other governmental charge, the depositary may (i)
deduct the amount thereof from any cash distributions, or (ii) sell deposited securities (by public or private sale) and
deduct the amount owing from the net proceeds of such sale. In either case, the ADR holder remains liable for any
shortfall. If any tax or governmental charge is unpaid, the depositary may also refuse to effect any registration,
registration of transfer, split up or combination of ADRs or withdrawal of deposited securities until such payment is made.
If any tax or governmental charge is required to be withheld on any cash distribution, the depositary may deduct the
amount required to be withheld from any cash distribution or, in the case of a non-cash distribution, sell the distributed
property or securities (by public or private sale) in such amounts and in such manner as the depositary deems necessary
and practicable to pay such taxes and distribute any remaining net proceeds or the balance of any such property after
deduction of such taxes to the ADR holders entitled thereto. Neither we nor the depositary nor any of our or its
respective agents, shall be liable to ADR holders or beneficial owners of the ADSs for failure of any of them to comply
with applicable tax laws, rules and/or regulations.
As an ADR holder or beneficial owner, you will be agreeing to indemnify us, the depositary, its custodian and any of our
or their respective officers, directors, employees, agents and affiliates against, and hold each of them harmless from, any
claims by any governmental authority with respect to taxes, additions to tax, penalties or interest arising out of any
refund of taxes, reduced rate of withholding at source or other tax benefit obtained, which obligations shall survive any
transfer or surrender of ADSs or the termination of the deposit agreement.
227
PART II
ITEM 13.DEFAULTS, DIVIDEND ARREARAGES AND DELINQUENCIES
None.
ITEM 14.MATERIAL MODIFICATIONS TO THE RIGHTS OF SECURITY
HOLDERS AND USE OF PROCEEDS
Not applicable.
ITEM 15.CONTROLS AND PROCEDURES
Disclosure Controls and Procedures
Our management, with the participation of our Group Chief Executive Officer and Group Chief Financial Officer, has
performed an evaluation of the effectiveness of our disclosure controls and procedures (as defined in Rules 13a-15(e)
and 15d-15(e) under the Exchange Act) as of December 31, 2025 and concluded that our disclosure controls and
procedures were not effective because of the material weakness in internal control over financial reporting described
below.
Notwithstanding management’s assessment that our disclosure controls and procedures were ineffective as of
December 31, 2025, our Group Chief Executive Officer and Group Chief Financial Officer have concluded that the
consolidated financial statements included in this Annual Report fairly present, in all material respects, our financial
position, results of operations and cash flows for the periods presented.
Management’s Annual Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal controls over financial reporting as
such term is defined in Rule 13a-15(f) and Rule 15d-15(f) under the Exchange Act.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatement. Also,
projections of any evaluation of the effectiveness of internal control to future periods are subject to the risk that controls
may become inadequate because of changes in conditions, or that the degree of compliance with the policies or
procedures may deteriorate.
Our management, with the participation of the Group Chief Executive Officer and Group Chief Financial Officer, has
evaluated the effectiveness of our internal controls over financial reporting as of December 31, 2025 based on the
criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring
Organizations of the Treadway Commission ("COSO").
Management has excluded RLS (USA) Inc. ("RLS") from its assessment of internal control over financial reporting as of
December 31, 2025, because it was acquired in a purchase business combination during 2025. RLS is a wholly-owned
subsidiary of the Company whose total assets and total sales excluded from management's assessment represent 34%
and 21% respectively, of each of the related consolidated financial statement amounts as of and for the year ended
December 31, 2025.
Based on this evaluation, our management, with the participation of our Group Chief Executive Officer and Group Chief
Financial Officer, has concluded that, as of December 31, 2025, our internal control over financial reporting was not
effective because of the material weakness described below.
A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that
there is a reasonable possibility that a material misstatement of its financial statements will not be prevented or detected
on a timely basis.
As previously disclosed in Item 15 of our Annual Report on Form 20-F for the year ended December 31, 2024, we
identified a material weakness arising from segregation of duties, which have not been sufficiently established across the
key business and financial processes to maintain appropriate segregation of duties over certain manual and information
technology business controls. This material weakness did not result in a misstatement of our annual consolidated
financial statements. However, the material weakness could result in a misstatement of our financial statement accounts
or disclosures that would result in a material misstatement to our annual consolidated financial statements that would not
be prevented or detected on a timely basis.
228
The effectiveness of the Company’s internal control over financial reporting as of December 31, 2025, has been audited
by PricewaterhouseCoopers, an independent registered public accounting firm as stated in their report, which appears in
the Consolidated financial statements on Form 20-F.
Plan for Remediation of Material Weakness in Internal Control over Financial Reporting
Management has initiated remediation actions, including redesigning roles and access rights to enforce segregation of
duties, implementing system-based workflow approvals, and enhancing documentation of review controls. We expect
these actions to be completed and tested for effectiveness during 2026.
Although we have made considerable progress and intend to complete these remediation activities, we will not be able to
fully remediate the material weakness until these steps have been completed, the enhanced processes have been
operating effectively for a sufficient period of time and appropriate testing has been performed. We provide no
assurances with respect to the timeline for implementing effective remedial measures, and our initiatives may not prove
to be successful in remediating the material weakness or preventing additional material weaknesses or significant
deficiencies in our internal control over financial reporting in the future.
Remediation of Previously Reported Material Weakness in Internal Control Over Financial
Reporting
As previously disclosed in Item 15 of our Annual Report on Form 20-F for the year ended December 31, 2024, we
identified a material weakness related to a lack of appropriately designed, implemented and documented procedures and
controls at both the entity-level and process-level to allow us to achieve complete, accurate and timely financial
reporting.
Since the material weakness has been identified, we have enhanced risk and control documentation practices related to
internal control over financial reporting, strengthened the monitoring and management testing of controls and oversight
mechanisms to promote ongoing compliance with internal control policies and procedures, continued the annual
Sarbanes-Oxley training program, and developed and implemented additional controls and procedures, including
compensating controls where necessary. These enhanced processes, including the implementation of new mitigating
controls, have now operated for a sufficient period of time and we have concluded, through testing, that they are
designed and are operating effectively. As a result, we have concluded the material weakness has been remediated as of
December 31, 2025.
Changes in Internal Control over Financial Reporting
Except as described above, there were no changes in our internal control over financial reporting during the period
covered by this Annual Report that have materially affected, or are reasonably likely to materially affect, our internal
control over financial reporting.
ITEM 16.RESERVED
ITEM 16A.AUDIT COMMITTEE FINANCIAL EXPERT
Jann Skinner is a member of our board of directors and serves on our Audit and Risk Committee as Chair. Our board of
directors has determined that Ms. Skinner is an audit committee financial expert and satisfies the “independence”
requirements of the rules and regulations of the SEC, the Nasdaq Listing Rules and the ASX Listing Rules.
ITEM 16B.CODE OF ETHICS
We have adopted a Code of Conduct applicable to all of our directors, officers, employees, consultants and contractors
to the Telix Group. Our Code of Conduct is publicly available on our website at www.telixpharma.com. We post on our
website all disclosures that are required by law, the ASX Listing Rules or the Nasdaq Listing Rules concerning any
amendments to, or waivers from, any provision of the Code of Conduct.
ITEM 16C.PRINCIPAL ACCOUNTANT FEES AND SERVICES
For the years ended December 31, 2025 and 2024, PricewaterhouseCoopers served as our independent registered
public accounting firm. The address for PricewaterhouseCoopers is 2 Riverside Quay, Southbank, Victoria 3006,
Australia.
229
The table below sets out the total amount of fees for services rendered to us by PricewaterhouseCoopers in the years
ended December 31, 2025 and 2024, and breaks down these amounts by category of service:
2025 | 2024 | |
US$ | US$ | |
(in thousands) | ||
Audit fees1 | 2,299 | 2,884 |
Audit-related fees2 | 86 | – |
Tax fees3 | – | 83 |
All other fees4 | 60 | – |
Total | 2,446 | 2,967 |
1.“Audit fees” include fees for services performed by our external auditor in connection with our annual audit for 2025
and 2024, fees related to the review of semi-annual financial statements, fees related to the pro forma financial
information, comfort letters, consents, and services that are normally provided in connection with statutory and
regulatory filings.
2.“Audit-related fees” relate to assurance and associated services that are traditionally performed by an independent
auditor, including accounting consultation and consultation concerning financial accounting, reporting standards and
due diligence.
3.“Tax fees” include fees for professional services rendered by our independent registered public accounting firm for
tax compliance, transfer pricing and tax advice on actual or contemplated transactions.
4.“All other fees” include fees for services rendered by our independent registered public accounting firm with respect
to any other advisory matters.
Pre-Approval Policies and Procedures
The Audit and Risk Committee has adopted a pre-approval policy for the engagement of our external auditor to perform
certain audit and non-audit services. Pursuant to this policy, which is designed to ensure that such engagements do not
impair the independence of our external auditor, pre-approval by the Audit and Risk Committee is required for all audit
and non-audit services to be performed by our external auditor, subject to the de minimis exceptions for non-audit
services described in Section 10A(i)(1)(B) of the Exchange Act which are approved by the Audit and Risk Committee prior
to the completion of the services.
All engagements by us of our external auditor for 2025 and 2024 were pre-approved by our Audit and Risk Committee.
ITEM 16D.EXEMPTIONS FROM LISTING STANDARDS FOR AUDIT
COMMITTEES
None.
ITEM 16E.PURCHASE OF EQUITY SECURITIES BY ISSUER AND AFFILIATED
PURCHASERS
None.
ITEM 16F.CHANGE IN REGISTRANT’S CERTIFYING ACCOUNTANT
None.
ITEM 16G.CORPORATE GOVERNANCE
Under Nasdaq Rule 5615(a)(3), foreign private issuers, such as our company, are permitted to follow certain home
country corporate governance practices instead of certain provisions of the Nasdaq rules. A foreign private issuer that
elects to follow a home country practice instead of any such Nasdaq rules must submit to Nasdaq, in advance, a written
statement from an independent counsel in such issuer’s home country certifying that the issuer’s practices are not
prohibited by the home country’s laws. We submitted such a written statement to Nasdaq. See “Item 6. Directors, Senior
Management and Employees—C. Board Practices— Foreign Private Issuer Exemption” for a summary of such
differences.
230
ITEM 16H.MINE SAFETY DISCLOSURE
None.
ITEM 16I.DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT
PREVENT INSPECTIONS
None.
ITEM 16J.INSIDER TRADING POLICIES
We have a comprehensive Securities Dealing Policy ("SDP"), which applies to all directors, officers and employees as well
as contractors and consultants whose terms of engagement apply the SDP to them. The SDP is reviewed and updated by
our board of directors on an as needed basis, and at least annually, for currency of practice. The purpose of the SDP is to
inform all persons to whom the SDP applies of the laws relating to insider trading, including pursuant to ASX Listing
Rules, the rules and regulations of the SEC, and the Nasdaq Listing Rules, and to provide them with practical guidance for
avoiding unlawful transactions in Telix securities and to protect our reputation.
Our SDP also prohibits short-term or speculative dealing in Telix securities by directors and employees. In addition,
directors and employees are not permitted to enter into any hedging arrangements that operate to limit the economic
risk associated with holding Telix securities or margin loan arrangements in relation to Telix securities. This includes
securities awarded under Telix’s equity incentive schemes.
A copy of the current SDP is available in the Corporate Governance section of our website at www.telixpharma.com, and
is included as an exhibit to this Annual Report.
ITEM 16K.CYBERSECURITY
frameworks to respond to and assess internal and external threats to the confidentiality, availability and integrity of our
data and information systems. Our cybersecurity policies, standards, processes, and practices are part of our information
security management system ("ISMS") program, which is aligned to ISO 27001:2022, an international standard for
information security management.
for assessing and managing our material risks from cybersecurity threats. The team is primarily responsible for our
overall cybersecurity risk management program and supervises both our internal cybersecurity personnel and our
external cybersecurity partners. Our cybersecurity management team has relevant academic backgrounds and
possesses extensive knowledge in cybersecurity risk management.
service providers by conducting vendor diligence. Vendors are generally assessed for risk based on the nature of their
service, access to data and systems and supply chain risk
threats that have materially affected or are reasonably likely to materially affect us, our business strategy, results of
operations, or financial condition.
Governance
and Risk Committee has responsibility for the oversight of cybersecurity, including the responsibility to review and
discuss with management and the Company’s auditors, as appropriate, management risks relating to data privacy,
any material cybersecurity incidents that may adversely affect the Company and on cybersecurity risks in general.
Group Chief Financial Officer. Our Chief Information Officer reports regularly to the Audit and Risk Committee and such
reporting includes topics such as risk assessments, risk management and control decisions, service provider
arrangements, test results, security incidents and responses and recommendations for changes and updates to policies
and procedures.
231
PART III
ITEM 17.FINANCIAL STATEMENTS
We have elected to include financial statements and related information specified in Item 18.
ITEM 18.FINANCIAL STATEMENTS
Financial statements are filed as part of this Annual Report, beginning on page F-1.
ITEM 19.EXHIBITS
The following documents are filed as part of this Annual Report.
Exhibit Number | Description of Exhibit |
Certificate of Registration of the Company (incorporated by reference to Exhibit 1.1 to the Company’s Registration Statement on Form 20-F (001-42128) filed October 30, 2024). | |
Constitution of the Company (incorporated by reference to Exhibit 1.2 to the Company’s Registration Statement on Form 20-F (001-42128) filed October 30, 2024). | |
Deposit Agreement (incorporated by reference to Exhibit 2.1 to the Company’s Registration Statement on Form 20-F (001-42128) filed October 30, 2024). | |
Form of American Depositary Receipt evidencing American Depositary Shares (included in Exhibit 2.1). | |
Description of Securities Registered under Section 12 of the Exchange Act (incorporated by reference to Exhibit 2.3 to the Company’s Annual Report on Form 20-F filed February 24, 2025). | |
License Agreement between Telix International Pty Ltd. and Eli Lilly Kinsale Limited, dated as of April 8, 2022, as amended (incorporated by reference to Exhibit 4.1 to the Company’s Registration Statement on Form 20-F (001-42128) filed October 30, 2024). | |
License Agreement between Telix International Pty Ltd. and Wilex AG, dated as of January 16, 2017, as amended (incorporated by reference to Exhibit 4.2 to the Company’s Registration Statement on Form 20-F (001-42128) filed October 30, 2024). | |
Form of Deed of Indemnity, Insurance and Access (filed herewith). | |
Lease Agreement, dated November 30, 2022, by and between Collan Investment Limited and Telix International Pty Ltd (incorporated by reference to Exhibit 4.4 to the Company’s Registration Statement on Form 20-F (001-42128) filed October 30, 2024). | |
Loan Agreement, dated March 3, 2022, by and between Telix Pharmaceuticals (Belgium) SPRL and BNP Paribas Fortis (incorporated by reference to Exhibit 4.6 to the Company’s Registration Statement on Form 20-F (001-42128) filed October 30, 2024). | |
Loan Agreement, dated March 3, 2022, by and between Telix Pharmaceuticals (Belgium) SPRL and IMBC (incorporated by reference to Exhibit 4.7 to the Company’s Registration Statement on Form 20-F (001-42128) filed October 30, 2024). | |
Equity Incentive Plan Rules (incorporated by reference to Exhibit 4.8 to the Company’s Annual Report on Form 20-F filed February 24, 2025). | |
Employment Agreement, dated January 16, 2017, by and between Telix Pharmaceuticals Limited and Christian Behrenbruch (filed herewith). | |
Employment Agreement, dated August 1, 2022, by and between Telix Pharmaceuticals Limited and Darren Smith (filed herewith). | |
Employment Agreement, dated December 20, 2023, by and between Telix Pharmaceuticals Limited and David Cade (filed herewith). | |
Employment Agreement, dated March 5, 2024, by and between Telix Pharmaceuticals (US) Inc. and Darren Patti (filed herewith). | |
Form of Non-Executive Director Agreement (incorporated by reference to Exhibit 4.13 to the Company’s Registration Statement on Form 20-F (001-42128) filed October 30, 2024). |
232
Agreement and Plan of Merger, dated as of February 7, 2024, by and among Telix Pharmaceuticals Limited, QSAM Biosciences, Inc., Cyclone Merger Sub I, Inc., Cyclone Merger Sub II, Inc. and David H. Clarke (incorporated by reference to Exhibit 4.14 to the Company’s Registration Statement on Form 20-F (001-42128) filed October 30, 2024). | |
Share Purchase Agreement, dated as of March 4, 2024, between ARTMS Inc. and Telix Pharmaceuticals Limited (incorporated by reference to Exhibit 4.15 to the Company’s Registration Statement on Form 20-F (001-42128) filed October 30, 2024). | |
Trust Deed, dated as of July 30, 2024, between Telix Pharmaceuticals Limited and The Hongkong and Shanghai Banking Corporation Limited (incorporated by reference to Exhibit 4.16 to the Company’s Registration Statement on Form 20-F (001-42128) filed October 30, 2024). | |
Stock Purchase Agreement, dated as of September 20, 2024, by and among Telix Pharmaceuticals (US) Inc., RLS Group Ltd., RLS (USA) Inc. and Perceptive Credit Holdings III, LP (incorporated by reference to Exhibit 4.17 to the Company’s Registration Statement on Form 20-F (001-42128) filed October 30, 2024). | |
Amendment No. 1 to the Stock Purchase Agreement, dated as of January 26, 2025, by and among Telix Pharmaceuticals (US) Inc., RLS Group Ltd., RLS (USA) Inc. and Perceptive Credit Holdings III, LP (incorporated by reference to Exhibit 4.18 to the Company’s Annual Report on Form 20-F filed February 24, 2025). | |
US Employee Stock Purchase Program (incorporated by reference to Exhibit 4.19 to the Company’s Annual Report on Form 20-F filed February 24, 2025). | |
Form of Deed of Indemnity and Insurance (filed herewith). | |
List of subsidiaries (filed herewith). | |
Securities Dealing Policy (filed herewith). | |
Certification of Principal Executive Officer pursuant to Rule 13a-14(a)/15d-14(a), as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 (filed herewith). | |
Certification of Principal Financial Officer pursuant to Rule 13a-14(a)/15d-14(a), as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 (filed herewith). | |
Certification of Principal Executive Officer pursuant to 18 U.S.C. §1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 (filed herewith). | |
Certification of Principal Financial Officer pursuant to 18 U.S.C. §1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 (filed herewith). | |
Consent of PricewaterhouseCoopers, independent registered public accounting firm (filed herewith). | |
Auditor's Independence Declaration | |
Independent Auditor's Report | |
Clawback / Dodd-Frank Compensation Recovery Policy (included as an Annexure to Exhibit 4.7 herein). | |
Appendix 4E | |
101.INS | Inline XBRL Instance Document. |
101.SCH | Inline XBRL Taxonomy Extension Schema Document |
101.CAL | Inline XBRL Taxonomy Extension Calculation Linkbase Document |
101.DEF | Inline XBRL Taxonomy Extension Definition Linkbase Document |
101.LAB | Inline XBRL Taxonomy Extension Label Linkbase Document |
101.PRE | Inline XBRL Taxonomy Extension Presentation Linkbase Document |
104 | Cover page Interactive Data File (embedded within the Inline XBRL document) |
+ Indicates management contract or compensatory plan.
233
SIGNATURES
The registrant hereby certifies that it meets all of the requirements for filing on Form 20-F and that it has duly caused
and authorized the undersigned to sign this annual report on its behalf.
TELIX PHARMACEUTICALS LIMITED | ||
By: | ||
Name: | Dr. Christian Behrenbruch | |
Title: | Managing Director and Group Chief Executive Officer | |
Date: February 20, 2026 | ||
F-1
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
Consolidated Financial Statements as of December 31, 2025 and 2024 and for the years ended
December 31, 2025, 2024 and 2023:
Page | |
( | |
F-2
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Shareholders of Telix Pharmaceuticals Limited
Opinions on the Financial Statements and Internal Control over Financial Reporting
We have audited the accompanying consolidated statement of financial position of Telix Pharmaceuticals Limited and its
subsidiaries (the “Company”) as of December 31, 2025 and 2024, and the related consolidated statements of
comprehensive income or loss, changes in equity and cash flows for each of the three years in the period ended
December 31, 2025, including the related notes (collectively referred to as the “consolidated financial statements”). We
also have audited the Company's internal control over financial reporting as of December 31, 2025, based on criteria
established in Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of
the Treadway Commission (COSO).
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial
position of the Company as of December 31, 2025 and 2024, and the results of its operations and its cash flows for each
of the three years in the period ended December 31, 2025 in conformity with International Financial Reporting Standards
as issued by the International Accounting Standards Board and Australian Accounting Standards and Interpretations as
issued by the Australian Accounting Standards Board. Also in our opinion, the Company did not maintain, in all material
respects, effective internal control over financial reporting as of December 31, 2025, based on criteria established in
Internal Control - Integrated Framework (2013) issued by the COSO because a material weakness in internal control over
financial reporting existed as of that date related to segregation of duties, which have not been sufficiently established
across the key business and financial processes to maintain appropriate segregation of duties over certain manual and
information technology business controls.
A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting, such
that there is a reasonable possibility that a material misstatement of the annual or interim financial statements will not be
prevented or detected on a timely basis. The material weakness referred to above is described in Management’s Annual
Report on Internal Control Over Financial Reporting appearing under Item 15. We considered this material weakness in
determining the nature, timing, and extent of audit tests applied in our audit of the December 31, 2025 consolidated
financial statements, and our opinion regarding the effectiveness of the Company’s internal control over financial
reporting does not affect our opinion on those consolidated financial statements.
Basis for Opinions
The Company's management is responsible for these consolidated financial statements, for maintaining effective internal
control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting
included in management's report referred to above. Our responsibility is to express opinions on the Company’s
consolidated financial statements and on the Company's internal control over financial reporting based on our audits. We
are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB)
and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and
the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and
perform the audits to obtain reasonable assurance about whether the consolidated financial statements are free of
material misstatement, whether due to error or fraud, and whether effective internal control over financial reporting was
maintained in all material respects.
Our audits of the consolidated financial statements included performing procedures to assess the risks of material
misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that
respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and
disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used
and significant estimates made by management, as well as evaluating the overall presentation of the consolidated
financial statements. Our audit of internal control over financial reporting included obtaining an understanding of internal
control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design
and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such
other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable
basis for our opinions.
As described in Management’s Annual Report on Internal Control Over Financial Reporting, management has excluded
RLS (USA) Inc. from its assessment of internal control over financial reporting as of December 31, 2025 because it was
acquired by the Company in a purchase business combination during 2025. We have also excluded RLS (USA) Inc. from
our audit of internal control over financial reporting. RLS (USA) Inc. is a wholly-owned subsidiary whose total assets and
total sales excluded from management’s assessment and our audit of internal control over financial reporting represent
34% and 21%, respectively, of the related consolidated financial statement amounts as of and for the year ended
December 31, 2025.
F-3
Definition and Limitations of Internal Control over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding
the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with
generally accepted accounting principles. A company’s internal control over financial reporting includes those policies
and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the
transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are
recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting
principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of
management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely
detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on
the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements.
Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become
inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may
deteriorate.
Critical Audit Matters
The critical audit matters communicated below are matters arising from the current period audit of the consolidated
financial statements that were communicated or required to be communicated to the audit committee and that (i) relate
to accounts or disclosures that are material to the consolidated financial statements and (ii) involved our especially
challenging, subjective, or complex judgments. The communication of critical audit matters does not alter in any way our
opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit
matters below, providing separate opinions on the critical audit matters or on the accounts or disclosures to which they
relate.
Acquisition of RLS (USA) Inc. – Valuation of Customer Relationships and Brand Name
As described in Note 23.1 to the consolidated financial statements, on January 28, 2025, the Company completed the
acquisition of RLS (USA) Inc. (“RLS”) for consideration of $240.9 million. Of the acquired intangible assets, $60.4 million
of customer relationships and $29.8 million of a brand name were recorded. Fair value is estimated by management
using a multi-period excess earnings method for customer relationships and a relief from royalty method for the brand
name. Management’s cash flow projections for the intangible assets acquired included significant judgments and
assumptions relating to revenue growth rates, earnings before interest and taxes (“EBIT”) margins, and discount rate for
customer relationships and revenue growth rates, royalty rate, and discount rate for the brand name.
The principal considerations for our determination that performing procedures relating to the valuation of customer
relationships and brand name acquired in the acquisition of RLS is a critical audit matter are (i) the significant judgment
by management when developing the fair value estimate of the customer relationships and brand name acquired; (ii) a
high degree of auditor judgment, subjectivity, and effort in performing procedures and evaluating management’s
significant assumptions related to revenue growth rates, EBIT margins, and discount rate for customer relationships and
revenue growth rates, royalty rate, and discount rate for the brand name; and (iii) the audit effort involved the use of
professionals with specialized skill and knowledge.
Addressing the matter involved performing procedures and evaluating audit evidence in connection with forming our
overall opinion on the consolidated financial statements. These procedures included testing the effectiveness of controls
relating to the acquisition accounting, including controls over management’s valuation of the customer relationships and
brand name acquired. These procedures also included, among others (i) reading the share purchase agreement; (ii)
testing management’s process for developing the fair value estimate of the customer relationships and brand name
acquired; (iii) evaluating the appropriateness of the multi-period excess earnings and relief from royalty methods used by
management; (iv) testing the completeness and accuracy of the underlying data used in the multi-period excess
earnings and relief from royalty methods; and (v) evaluating the reasonableness of the significant assumptions used by
management related to revenue growth rates, EBIT margins, and discount rate for customer relationships and revenue
growth rates, royalty rate, and discount rate for the brand name. Evaluating management’s assumptions related to
revenue growth rates and EBIT margins for customer relationships and revenue growth rates for the brand name
involved considering: (i) the performance of the RLS business; (ii) the consistency with external market and industry
data; and (iii) whether the assumptions were consistent with evidence obtained in other areas of the audit. Professionals
with specialized skill and knowledge were used to assist in evaluating: (i) the appropriateness of the multi-period excess
earnings and relief from royalty methods; and (ii) the reasonableness of the revenue growth rates, EBIT margins, and
discount rate for customer relationships and revenue growth rates, royalty rate, and discount rate for the brand name.
Impairment Assessment of Indefinite-Lived Intangible Assets and Goodwill – TLX090-Tx and
ARTMS Cash-Generating Units
As described in Note 21 to the consolidated financial statements, the Company’s intangible assets balance including
goodwill was $592.8 million as of December 31, 2025. The carrying amount associated with the TLX090-Tx and ARTMS
cash-generating units, which includes indefinite-lived intangible assets and goodwill, was $92.8 million and $85.2 million
as of December 31, 2025, respectively. Management conducts an impairment test as of December 31 of each year, or
F-4
more frequently if events or circumstances indicate the carrying value of cash-generating units containing indefinite-
lived intangible assets and goodwill may be impaired. Potential impairment is identified by comparing the fair value of a
cash-generating unit to its carrying value, including goodwill. Fair value is estimated by management using a discounted
cash flow model. Management’s cash flow projections for the TLX090-Tx and ARTMS included significant judgments and
assumptions relating to expected sales volumes, net sales prices, probability of success, terminal value, and discount
rates.
The principal considerations for our determination that performing procedures relating to the indefinite-lived intangible
assets and goodwill impairment assessments of the TLX090-Tx and ARTMS cash-generating units is a critical audit
matter are (i) the significant judgment by management when developing the fair value estimate of the TLX090-Tx and
ARTMS cash-generating units; (ii) a high degree of auditor judgment, subjectivity, and effort in performing procedures
and evaluating management’s significant assumptions related to expected sales volumes, net sales prices, probability of
success, terminal value, and discount rates; and (iii) the audit effort involved the use of professionals with specialized
skill and knowledge.
Addressing the matter involved performing procedures and evaluating audit evidence in connection with forming our
overall opinion on the consolidated financial statements. These procedures included testing the effectiveness of controls
relating to management’s indefinite-lived intangible assets and goodwill impairment assessments, including controls over
the valuation of the Company’s TLX090-Tx and ARTMS cash-generating units. These procedures also included, among
others (i) testing management’s process for developing the fair value estimate of the TLX090-Tx and ARTMS cash-
generating units; (ii) evaluating the appropriateness of the discounted cash flow model used by management; (iii) testing
the completeness and accuracy of underlying data used in the discounted cash flow model; and (iv) evaluating the
reasonableness of the significant assumptions used by management related to expected sales volumes, net sales prices,
probability of success, terminal value, and discount rates. Evaluating management’s assumptions related to expected
sales volumes, net sales prices, probability of success, and terminal value involved evaluating whether the assumptions
used by management were reasonable considering (i) the performance of the TLX090-Tx and ARTMS cash-generating
units; (ii) the consistency with external market and industry data; and (iii) whether the assumptions were consistent with
evidence obtained in other areas of the audit. Professionals with specialized skill and knowledge were used to assist in
evaluating (i) the appropriateness of the discounted cash flow model; and (ii) the reasonableness of the discount rates
assumptions.
/s/ PricewaterhouseCoopers
Melbourne, Australia
February 20, 2026
We have served as the Company’s auditor since 2017.
F-5
Consolidated statement of comprehensive income or loss for the year ended December 31, 2025
2025 | 2024 | 2023 | |||||
Note | US$'000 | US$'000 | US$'000 | ||||
(Recast) | (Recast) | ||||||
Continuing operations | |||||||
Revenue from contracts with customers | 4 | ||||||
Cost of sales | ( | ( | ( | ||||
Gross profit | |||||||
Research and development costs | 5 | ( | ( | ( | |||
Selling and marketing expenses | ( | ( | ( | ||||
Manufacturing and distribution costs | 6 | ( | ( | ( | |||
General and administration costs | 7 | ( | ( | ( | |||
Other gains/(losses)(net) | 10 | ( | |||||
Operating profit | |||||||
Finance income | |||||||
Finance costs | 11 | ( | ( | ( | |||
(Loss)/profit before income tax | ( | ||||||
Income tax (expense)/benefit | 12 | ( | ( | ||||
(Loss)/profit for the year | ( | ||||||
(Loss)/profit for the year attributable to: | |||||||
Owners of Telix Pharmaceuticals Limited | ( | ||||||
Other comprehensive (loss)/income: | |||||||
Items that will not be reclassified to profit or loss in subsequent periods: | |||||||
Changes in the fair value of investments at fair value through other comprehensive income | ( | ( | ( | ||||
Items to be reclassified to profit or loss in subsequent periods: | |||||||
Exchange differences on translation of foreign operations | ( | ( | |||||
Total comprehensive (loss)/income for the year | ( | ||||||
Total comprehensive (loss)/income for the year attributable to: | |||||||
Owners of Telix Pharmaceuticals Limited | ( |
2025 | 2024 | 2023 | |||||
Note | Cents | Cents | Cents | 1. | |||
Basic (loss)/earnings per share from continuing operations after income tax attributable to the ordinary equity holders of the Company | 13.1 | ( | |||||
Diluted (loss)/earnings per share from continuing operations after income tax attributable to the ordinary equity holders of the Company | 13.2 | ( |
The above consolidated statement of comprehensive income or loss should be read in conjunction with the
accompanying notes.
F-6
Consolidated statement of financial position as at December 31, 2025
2025 | 2024 | 1 January 2024 | |||||
Note | US$'000 | US$'000 | US$'000 | ||||
(Recast) | (Recast) | ||||||
Current assets | |||||||
Cash and cash equivalents | |||||||
Trade and other receivables | 14 | ||||||
Inventories | 15 | ||||||
Current tax asset | |||||||
Other current assets | 16 | ||||||
Total current assets | |||||||
Non-current assets | |||||||
Financial assets | 17 | ||||||
Deferred tax assets | 18.1 | ||||||
Property, plant and equipment | 19 | ||||||
Right-of-use assets | 20 | ||||||
Intangible assets | 21 | ||||||
Other non-current assets | 22 | ||||||
Total non-current assets | |||||||
Total assets | |||||||
Current liabilities | |||||||
Trade and other payables | 24 | ||||||
Borrowings | 25 | ||||||
Current tax payable | |||||||
Contract liabilities | |||||||
Lease liabilities | 26 | ||||||
Provisions | 27 | ||||||
Contingent consideration | 28 | ||||||
Employee benefit obligations | 29 | ||||||
Total current liabilities | |||||||
Non-current liabilities | |||||||
Borrowings | 25 | ||||||
Contract liabilities | |||||||
Lease liabilities | 26 | ||||||
Deferred tax liabilities | 18.2 | ||||||
Other non-current liabilities | |||||||
Provisions | 27 | ||||||
Contingent consideration | 28 | ||||||
Employee benefit obligations | 29 | ||||||
Total non-current liabilities | |||||||
Total liabilities | |||||||
Net assets |
F-7
2025 | 2024 | 1 January 2024 | |||||
Note | US$'000 | US$'000 | US$'000 | ||||
(Recast) | (Recast) | ||||||
Equity | |||||||
Share capital | 30.1 | ||||||
Share capital reserve | 30.2 | ( | ( | ||||
Other reserves | 30.3 | ||||||
Accumulated losses | ( | ( | ( | ||||
Total equity |
The above consolidated statement of financial position should be read in conjunction with the accompanying notes.
F-8
Consolidated statement of changes in equity for the year ended December 31, 2025
Share capital | Share capital reserve | Other reserves | Accumulated losses | Total equity | |||||||
Note | US$'000 | US$'000 | US$'000 | US$'000 | US$'000 | ||||||
Balance as at January 1, 2025 | ( | ||||||||||
Loss for the year | - | - | - | ( | ( | ||||||
Other comprehensive loss | - | - | ( | - | ( | ||||||
Total comprehensive loss | - | - | ( | ( | ( | ||||||
Issue of shares on acquisitions | 30.1 | - | - | - | |||||||
Issue of shares on exercise of options | 30.1, 30.2 | ( | - | - | |||||||
Share-based payments to employees | 30.3 | - | - | - | |||||||
Share-based payments associated with acquisitions | 30.3 | - | - | - | |||||||
Transfer on satisfaction of acquisition performance rights | 30.1, 30.3 | - | ( | - | |||||||
Transfer on exercise of options | 30.3 | - | - | ( | |||||||
( | |||||||||||
Balance as at December 31, 2025 | ( | ( | |||||||||
(Recast) | (Recast) | (Recast) | (Recast) | (Recast) | |||||||
Balance as at January 1, 2024 | ( | ( | |||||||||
Profit for the year | - | - | - | ||||||||
Other comprehensive income | - | - | - | ||||||||
Total comprehensive income | - | - | |||||||||
Issue of shares on acquisitions | 30.1 | - | - | - | |||||||
Issue of shares on exercise of options | 30.1, 30.2 | ( | - | - | |||||||
Issue of convertible bonds | |||||||||||
Transaction costs arising on convertible bonds issue | ( | ( | |||||||||
Share-based payments to employees | 30.3 | - | - | - | |||||||
Share-based payments associated with acquisitions | 30.3 | - | - | - | |||||||
Transfer on exercise of options | 30.3 | - | - | ( | |||||||
Balance as at December 31, 2024 | ( |
F-9
Share capital | Share capital reserve | Other reserves | Accumulated losses | Total equity | |||||||
Note | US$'000 | US$'000 | US$'000 | US$'000 | US$'000 | ||||||
(Recast) | (Recast) | (Recast) | (Recast) | (Recast) | |||||||
Balance as at January 1, 2023 | ( | ( | |||||||||
Profit for the year | - | - | - | ||||||||
Other comprehensive loss | - | - | ( | - | ( | ||||||
Total comprehensive income | - | - | ( | ||||||||
Issue of shares on acquisitions | 30.1 | - | - | - | |||||||
Issue of shares on exercise of options | 30.1, 30.2 | ( | - | - | |||||||
Share-based payments to employees | 30.3 | - | - | - | |||||||
Share-based payments associated with acquisitions | 30.3 | - | - | - | |||||||
Transfer on exercise of options | 30.3 | - | - | ( | |||||||
( | |||||||||||
Balance as at December 31, 2023 | ( | ( |
The above consolidated statement of changes of equity should be read in conjunction with the accompanying notes.
F-10
Consolidated statement of cash flows for the year ended December 31, 2025
2025 | 2024 | 2023 | ||||||
Note | US$'000 | US$'000 | US$'000 | |||||
(Recast) | (Recast) | |||||||
Cash flows from operating activities | ||||||||
Receipts from customers | ||||||||
Payments to suppliers and employees | ( | ( | ( | |||||
Payments for contingent consideration | ( | ( | ( | |||||
Income taxes paid | ( | ( | ( | |||||
Interest received | ||||||||
Interest paid | ( | ( | ( | |||||
Net cash (used in)/from operating activities | 32.1 | ( | ||||||
Cash flows from investing activities | ||||||||
Payments for investments in financial assets | ( | ( | ( | |||||
Payments for acquisition of subsidiaries, net of cash acquired | ( | ( | ||||||
Purchases of intangible assets | ( | ( | ( | |||||
Purchases of other non-current assets | ( | ( | ||||||
Purchases of property, plant and equipment | ( | ( | ( | |||||
Payments for contingent consideration | ( | ( | ( | |||||
Payments for deferred consideration | ( | |||||||
Payments for decommissioning liability | ( | |||||||
Net cash used in investing activities | ( | ( | ( | |||||
Cash flows from financing activities | ||||||||
Proceeds from borrowings | ||||||||
Repayment of borrowings | ( | ( | ||||||
Principal element of lease payments | ( | ( | ( | |||||
Proceeds from issue of shares and other equity | ||||||||
Transaction costs of borrowings or capital raising | ( | |||||||
Net cash (used in)/provided by financing activities | ( | |||||||
Net (decrease)/increase in cash held | ( | |||||||
Net foreign exchange differences | ( | |||||||
Cash and cash equivalents at the beginning of the financial year | ||||||||
Cash and cash equivalents at the end of the financial year |
The above consolidated statement of cash flows should be read in conjunction with the accompanying notes.
F-11
Notes to the consolidated financial statements
1. Corporate information
Telix Pharmaceuticals Limited ("Telix" or "the Company") is a for-profit company incorporated and domiciled in Australia.
It is limited by shares that are publicly traded on the Australian Securities Exchange (ASX: TLX) and on the Nasdaq Global
Select Market (NASDAQ: TLX). These consolidated financial statements comprise the results of Telix and its subsidiaries
(together referred to as "the Group"). The consolidated financial statements were authorized for issue in accordance with
a resolution of the Directors on February 20, 2026.
2. Material accounting policy information
The material accounting policies that have been used in the preparation of these financial statements are summarized
below.
2.1. Going concern
The Directors are satisfied that the Group continues to be a going concern as at the date of these financial statements.
Further, the Directors are of the opinion that no asset is likely to be realized for an amount less than the amount at which
it is recorded in the consolidated statement of financial position as at December 31, 2025.
As such, no adjustment has been made to the financial statements relating to the recoverability and classification of the
asset carrying amounts or the classification of liabilities that might be necessary should the Group not continue as a
going concern.
2.2. Basis of preparation
Telix Pharmaceuticals Limited is a for-profit entity for the purpose of preparing the financial statements.
These general purpose financial statements have been prepared in accordance with International Financial Reporting
Standards as issued by the International Accounting Standards Board ("IFRS Accounting Standards") and Australian
Accounting Standards and Interpretations as issued by the Australian Accounting Standards Board and the Australian
Corporations Act 2001 (Cth).
The financial statements have been prepared on a historical cost basis, except for certain financial instruments, which
have been measured at fair value.
1.Comparatives
Where necessary, comparative information has been re-classified to achieve consistency in disclosure with current
financial amounts and other disclosures.
2.New and amended standards adopted by the Group
The Group has adopted all relevant new and amended standards and interpretations issued by the International
Accounting Standards Board which are effective for annual reporting periods beginning on January 1, 2025.
3.New standards and interpretations not yet adopted
Certain new accounting standards and interpretations have been published that are not mandatory for December 31,
2025 reporting periods and have not been early adopted by the Group.
4.IFRS 18 Presentation and Disclosure in Financial Statements (effective for annual periods
beginning on or after January 1, 2027)
IFRS 18 will replace IAS 1 Presentation of financial statements, introducing new requirements that will help to achieve
comparability of the financial performance of similar entities and provide more relevant information and transparency to
users. Even though IFRS 18 will not impact the recognition or measurement of items in the financial statements, its
impacts on presentation and disclosure are expected to be pervasive, in particular those related to the statement of
comprehensive income or loss and providing management-defined performance measures within the financial
statements.
Management is currently assessing the detailed implications of applying the new standard on the Group’s consolidated
financial statements.
2.3. Significant changes in the current reporting period
2.3.1 Change in presentation currency
The Group has retrospectively changed its presentation currency from Australian Dollars to United States Dollars (USD or
US$). The change in presentation currency, in the opinion of the Directors, will provide investors and other stakeholders
F-12
with a clearer and more reliable understanding of the Group’s business performance and is more comparable to the
Group’s peers, most of which are presented in U.S. Dollars.
The voluntary change in presentational currency is effective from 1 January 2025 and has been accounted for
retrospectively in the consolidated financial statements.
The financial report has been recast to USD using the procedures outlined below:
1.Consolidated statement of comprehensive income or loss and consolidated statement of cash flows have been
translated into USD using foreign currency rates at the dates of transactions prevailing for the relevant period;
2.Assets and liabilities in the consolidated statement of financial position have been translated into USD at the closing
foreign currency rates on the relevant reporting dates;
3.The equity section of the consolidated statement of financial position has been translated into USD as follows:
a.Foreign currency translation reserve, share based payment reserve and accumulated losses have been translated
to USD using foreign currency rates at the dates of transactions prevailing for the relevant period.
b.Share capital and share capital reserves have been translated into USD using historical rates;
4.Earnings per share has been translated into USD based on the translated USD net income, translated as described
above.
2.4. Principles of consolidation
Subsidiaries are all entities (including structured entities) over which the Group has control. The Group controls an entity
when the Group is exposed to, or has rights to, variable returns from its involvement with the entity and has the ability to
affect those returns through its power to direct the activities of the entity. Subsidiaries are fully consolidated from the
date on which control is transferred to the Group. If the Group loses control of a subsidiary, the Group derecognizes the
assets and liabilities of the former subsidiary from the consolidated statement of financial position and recognizes the
gain or loss associated with the loss of control attributable to the former controlling interest.
Intercompany transactions, balances and unrealized gains on transactions between Group companies are eliminated on
consolidation. Unrealized losses are also eliminated unless the transaction provides evidence of an impairment of the
transferred asset. Accounting policies of subsidiaries have been changed where necessary to ensure consistency with
the policies adopted by the Group.
2.5. Foreign currency translation
1.Functional and presentation currency
Items included in the financial statements of each of the Group’s entities are measured using the currency of the primary
economic environment in which the entity operates (the functional currency). The consolidated financial statements are
presented in U.S. Dollars.
2.Transactions and balances
Foreign currency transactions are translated into the functional currency using the exchange rates at the dates of the
transactions. Foreign exchange gains and losses resulting from the settlement of such transactions and from the
translation of monetary assets and liabilities denominated in foreign currencies at year end exchange rates are generally
recognized in profit or loss. Foreign exchange gains and losses that relate to borrowings are presented in the
consolidated statement of comprehensive income or loss, within finance costs. All other foreign exchange gains and
losses are presented in the consolidated statement of comprehensive income or loss on a net basis within other income
or other expenses.
Non-monetary items that are measured at fair value in a foreign currency are translated using the exchange rates at the
date when the fair value was determined.
Translation differences on assets and liabilities carried at fair value are reported as part of the fair value gain or loss.
3.Group companies
The results and financial position of foreign operations (none of which has the currency of a hyperinflationary economy)
that have a functional currency different from the presentation currency are translated into the presentation currency as
follows:
•assets and liabilities for each consolidated statement of financial position presented are translated at the closing
rate at the date of that consolidated statement of financial position
F-13
•income and expenses for each consolidated statement of comprehensive income or loss are translated at actual
exchange rates at the dates of the transactions, and
•all resulting exchange differences are recognized in other comprehensive income.
On consolidation, exchange differences arising from the translation of any net investment in foreign entities, and of
borrowings and other financial instruments designated as hedges of such investments, are recognized in other
comprehensive income. When a foreign operation is sold or any borrowings forming part of the net investment are
repaid, the associated exchange differences are reclassified to profit or loss, as part of the gain or loss on sale. Goodwill
and fair value adjustments arising on the acquisition of a foreign operation are treated as assets and liabilities of the
foreign operation and translated at the closing rate.
2.6. Business combinations
The acquisition method of accounting is used to account for all business combinations, regardless of whether equity
instruments or other assets are acquired. The consideration transferred for the acquisition of a subsidiary comprises the:
•fair values of the assets transferred
•liabilities incurred to the former owners of the acquired business
•equity interests issued by the Group
•fair value of any asset or liability resulting from a contingent consideration arrangement, and
•fair value of any pre-existing equity interest in the subsidiary.
Identifiable assets acquired and liabilities and contingent liabilities assumed in a business combination are, with limited
exceptions, measured initially at their fair values at the acquisition date. Acquisition-related costs are expensed as
incurred. The excess of the consideration transferred, amount of any non-controlling interest in the acquired entity, and
acquisition-date fair value of any previous equity interest in the acquired entity over the fair value of the net identifiable
assets acquired is recorded as goodwill. If those amounts are less than the fair value of the net identifiable assets of the
subsidiary acquired, the difference is recognized directly in profit or loss as a bargain purchase.
Where settlement of any part of cash consideration is deferred, the amounts payable in the future are discounted to their
present value as at the date of exchange. The post-tax discount rate used is the entity’s incremental borrowing rate,
being the rate at which a similar borrowing could be obtained from an independent financier under comparable terms and
conditions. Contingent consideration is classified either as equity or a financial liability. Amounts classified as a financial
liability are subsequently remeasured to fair value with changes in fair value recognized in profit or loss.
The acquisition date carrying value of the Group’s previously held equity interest in the acquiree is remeasured to fair
value at the acquisition date. Any gains or losses arising from such remeasurement are recognized in profit or loss. If the
initial accounting for a business combination is incomplete by the end of the reporting period in which the combination
occurs, the Group reports provisional amounts for the items for which the accounting is incomplete. Those provisional
amounts are adjusted during the measurement period (see below), or additional assets or liabilities are recognized, to
reflect new information obtained about facts and circumstances that existed as of the acquisition date that, if known,
would have affected the amounts recognized as of that date. The measurement period is the period from the date of
acquisition to the date the Group obtains complete information about facts and circumstances that existed as of the
acquisition date and is subject to a maximum of one year .
2.7. Current and non-current classification
Assets and liabilities are presented in the consolidated statement of financial position based on current and non- current
classification.
An asset is current when it is expected to be realized or intended to be sold or consumed in the Group’s normal operating
cycle; it is held primarily for the purpose of trading; it is expected to be realized within 12 months after the reporting
period; or the asset is cash or cash equivalent unless restricted from being exchanged or used to settle a liability for at
least 12 months after the reporting period. All other assets are classified as non-current.
A liability is current when it is expected to be settled in the Group’s normal operating cycle; it is held primarily for the
purpose of trading; it is due to be settled within 12 months after the reporting period; or there is no right to defer the
settlement of the liability for at least 12 months after the reporting period. All other liabilities are classified as non-current.
For instances where a liability is based on sales volumes, the payment expected to be realized within 12 months is
current based on the underlying estimate of the timing of sales.
Deferred tax assets and liabilities are always classified as non-current.
F-14
2.8. Cash and cash equivalents
For the purpose of presentation in the consolidated statement of cash flows, cash and cash equivalents includes cash on
hand, deposits held at call with financial institutions, other short-term, highly liquid investments with original maturities of
three months or less that are readily convertible to known amounts of cash and which are subject to an insignificant risk
of changes in value, and bank overdrafts. Bank overdrafts are shown within borrowings in current liabilities in the
consolidated statement of financial position.
2.9. Trade and other receivables
Trade receivables and other receivables are all classified as financial assets held at amortized cost. Trade receivables
are recognized initially at the amount of consideration that is unconditional, unless they contain significant financing
components when they are recognized at fair value.
Impairment of trade and other receivables
The collectability of trade and other receivables is reviewed on an ongoing basis. Individual debts which are known to be
uncollectible are written off when identified. The Group recognizes an impairment provision based upon anticipated
lifetime losses of trade receivables.
The anticipated losses are determined with reference to historical loss experience (when it is available) and are regularly
reviewed and updated. They are subsequently measured at amortized cost using the effective interest method, less loss
allowance. See note 33.4 for further information about the Group’s accounting for trade receivables and description of
the Group’s impairment policies.
2.10. Inventories
Raw materials and stores, work in progress and finished goods
Raw materials and stores, work in progress and finished goods are stated at the lower of cost and net realizable value.
Cost comprises direct materials, direct labor and an appropriate proportion of variable and fixed overhead expenditure,
the latter being allocated on the basis of normal operating capacity. Cost includes the reclassification from equity of any
gains or losses on qualifying cash flow hedges relating to purchases of raw material but excludes borrowing costs. Costs
are assigned to individual items of inventory on the basis of weighted average costs. Costs of purchased inventory are
determined after deducting rebates and discounts. Net realizable value is the estimated selling price in the ordinary
course of business less the estimated costs of completion and the estimated costs necessary to make the sale. Clinical
and pre-launch inventory with no alternative use is expensed as produced and recorded as research and development
expense.
2.11. Property, plant and equipment
All property, plant and equipment is stated at historical cost less accumulated depreciation. Historical cost includes
expenditure that is directly attributable to the acquisition of the items. Cost may also include transfer from equity of any
gains or losses on qualifying cash flow hedges of foreign currency purchases of property, plant and equipment.
Subsequent costs are included in the asset’s carrying amount or recognized as a separate asset, as appropriate, only
when it is probable that future economic benefits associated with the item will flow to the Group and the cost of the item
can be measured reliably.
The carrying amount of any component accounted for as a separate asset is derecognized when replaced. All other
repairs and maintenance are charged to profit or loss during the reporting period in which they are incurred.
Depreciation is calculated using the straight-line method to allocate the cost, net of the residual values, over the
estimated useful lives. The assets’ residual values and useful lives are reviewed, and adjusted if appropriate, at the end
of each reporting period. An asset’s carrying amount is written down immediately to its recoverable amount if the asset’s
carrying amount is greater than its estimated recoverable amount.
The useful lives of assets are as follows:
•Buildings: 18 years
•Plant and equipment: 3 -15 years
•Furniture, fittings and equipment: 3 -5 years
•Leased plant and equipment: 3 -5 years
Gains and losses on disposals are determined by comparing proceeds with carrying amount. These are included in profit
or loss. When revalued assets are sold, it is Group policy to transfer any amounts included in other reserves in respect of
those assets to accumulated losses.
F-15
2.12. Lease liabilities
Liabilities arising from a lease are initially measured on a present value basis. Lease liabilities include the net present
value of the following lease payments:
•fixed payments (including in-substance fixed payments), less any lease incentives receivable
•variable lease payments that are based on an index or a rate, initially measured using the index or rate as at the
commencement date
•amounts expected to be payable by the Group under residual value guarantees
•the exercise price of a purchase option if the Group is reasonably certain to exercise that option, and
•payments of penalties for terminating the lease, if the lease term reflects the Group exercising that option.
Lease payments to be made under reasonably certain extension options are also included in the measurement of the
liability.
Leases are recognized as a right-of-use asset and a corresponding liability at the date at which the leased asset is
available for use by the Group. Each lease payment is allocated between the liability and finance cost. The finance cost is
charged to profit or loss over the lease period so as to produce a constant periodic rate of interest on the remaining
balance of the liability for each period.
2.13. Right-of-use assets
Right-of-use assets are measured at cost comprising the following:
•the amount of the initial measurement of lease liability
•any lease payments made at or before the commencement date less any lease incentives received
•any initial direct costs, and
•restoration costs.
Right-of-use assets are depreciated over the shorter of the asset’s useful life and the lease term on a straight-line basis.
If the Group is reasonably certain to exercise a purchase option, the right-of-use asset is depreciated over the underlying
asset’s useful life.
2.14. Non-current financial assets
Non-current financial assets held for long-term strategic purposes are classified within non-current assets on the
consolidated statement of financial position. The financial impacts related to these financial assets are recorded in other
comprehensive income.
Non-current financial assets are initially recorded at fair value on their trade date, which is different from the settlement
date when the transaction is ultimately effected. Quoted securities are remeasured at each reporting date to fair value
based on current market prices. If the market for a financial asset is not active or no market is available, fair values are
established using valuation techniques.
Equity securities held as strategic investments are generally designated at the date of acquisition as financial assets
valued at fair value through other comprehensive income with no subsequent recycling through profit or loss. Unrealized
gains and losses, including exchange gains and losses, are recorded as a fair value adjustment in the consolidated
statement of comprehensive income. They are reclassified to retained earnings when the equity security is sold.
2.15. Intangible assets
1.Goodwill
Goodwill on acquisitions of subsidiaries is included in intangible assets. Goodwill is not amortized, but is tested for
impairment annually, or more frequently if events or changes in circumstances indicate that it might be impaired and is
carried at cost less accumulated impairment losses. Gains and losses on the disposal of an entity include the carrying
amount of goodwill relating to the entity sold. Goodwill is allocated to cash-generating units for the purpose of
impairment testing. The allocation is made to those cash-generating units or group of cash-generating units that are
expected to benefit from the business combination in which the goodwill arose.
2.Patents, trademarks, licenses and customer contracts
Separately acquired trademarks and licenses are shown at historical cost. Trademarks, licenses and customer contracts
acquired in a business combination are recognized at fair value at the acquisition date. They have a finite useful life and
F-16
are subsequently carried at cost less accumulated amortization and impairment losses. The useful life of these
intangibles assets is 5 to 20 years.
3.Intellectual property
Intellectual property arising from business combinations is recognized at fair value when separately identifiable from
goodwill. Intellectual property is recorded as an indefinite life asset when it is not yet ready for use. At the point the asset
is ready for use, the useful life is reassessed as a definite life asset and amortized over a period of 5 to 20 years.
Amortization and impairment charges related to currently marketed products are recognized in cost of goods sold.
Assets not available for use are tested annually for impairment. Assets are carried at cost less accumulated impairment
losses and/or accumulated amortization. An impairment trigger assessment is performed annually for assets available for
use.
4.Research and development
Research expenditure on internal projects is recognized as an expense as incurred. Costs incurred on development
projects (relating to the design and testing of new or improved products) are recognized as intangible assets when it is
probable that the project will, after considering its commercial and technical feasibility, be completed and generate
future economic benefits and its costs can be measured reliably. The expenditure that could be recognized comprises all
directly attributable costs, including costs of materials, services, direct labor and an appropriate proportion of overheads.
Other expenditures that do not meet these criteria are recognized as an expense as incurred. As the Group has not met
the requirement under the standard to recognize costs in relation to development as intangible assets, these amounts
have been expensed within the financial statements.
2.16. Impairment of assets
Goodwill and intangible assets that have an indefinite useful life are not subject to amortization and are tested annually
for impairment, or more frequently if events or changes in circumstances indicate that they might be impaired. Other
assets are tested for impairment whenever events or changes in circumstances indicate that the carrying amount may
not be recoverable. An impairment loss is recognized for the amount by which the asset’s carrying amount exceeds its
recoverable amount. The recoverable amount is the higher of an asset’s fair value less costs of disposal and value in use.
For the purposes of assessing impairment, assets are grouped at the lowest levels for which there are separately
identifiable cash inflows which are largely independent of the cash inflows from other assets or Groups of assets (cash-
generating units). Non-financial assets other than goodwill that suffered an impairment are reviewed for possible reversal
of the impairment at the end of each reporting period.
2.17. Trade and other payables
These amounts represent liabilities for goods and services provided to the Group prior to the reporting date which are
unpaid. The amounts are unsecured and are usually paid within 30 days of recognition. Trade and other payables are
presented as current liabilities unless payment is not due within 12 months after the reporting period. They are
recognized initially at fair value and subsequently measured at amortized cost using the effective interest method.
2.18. Provisions
Provisions are recognized when the Group has a present (legal or constructive) obligation as a result of a past event, it is
probable the Group will be required to settle the obligation, and a reliable estimate can be made of the amount of the
obligation. The amount recognized as a provision is the best estimate of the consideration required to settle the present
obligation at the reporting date, taking into account the risks and uncertainties surrounding the obligation. If the time
value of money is material, provisions are discounted using a current pre-tax rate specific to the liability. The increase in
the provision resulting from the passage of time is recognized as a finance cost.
Decommissioning liability
The Group has recognized a provision for its obligation to decommission its radiopharmaceutical production facility at the
end of its operating life. At the end of a facility’s life, costs are incurred in safely removing certain assets involved in the
production of radioactive isotopes. The Group recognizes the full discounted cost of decommissioning as an asset and
liability when the obligation to restore sites arises. The decommissioning asset is included within property, plant and
equipment with the cost of the related installation. The liability is included within provisions. Revisions to the estimated
costs of decommissioning which alter the level of the provisions required are also reflected in adjustments to the
decommissioning asset. The amortization of the asset is included in the consolidated statement of comprehensive
income or loss and the unwinding of discount of the provision is included within Finance costs. Further detail has been
provided in note 27.2.
2.19. Contingent consideration
The contingent consideration liabilities associated with business combinations are measured at fair value which has been
calculated with reference to our judgment of the expected probability and timing of the potential future milestone
payments, which is then discounted to a present value using appropriate discount rates with reference to the Group’s
F-17
weighted average cost of capital. Subsequent changes in estimates for contingent consideration liabilities are recognized
in Other gains/(losses) (net). The effect of unwinding the discount over time is recognized in Finance costs.
Contingent consideration in connection with the purchase of individual assets outside of business combinations is
recognized as a liability only when a non-contingent obligation arises (i.e. when milestone is met). Where the contingent
consideration is payable in shares, or the group has an election to pay in shares, it is accounted for as an equity settled
share-based payment. Equity settled share-based payments are recognized at their fair value at the date control of the
asset is obtained. The determination of whether the payment should be capitalized or expensed is usually based on the
reason for the contingent payment. If the contingent payment is based on regulatory approvals received (i.e.
development milestone), it will generally be capitalized as the payment is incidental to the acquisition so the asset may
be made available for its intended use. If the contingent payment is based on period volumes sold (i.e. sales related
milestone), it will generally be expensed.
Changes in the fair value of liabilities from contingent consideration will be capitalized or expensed based on the nature
of the asset acquired (refer above), except for the effect from unwinding discounts. Interest rate effects from unwinding
of discounts are recognized as finance costs. The fair value of equity-settled share-based payments is not re-assessed
once the asset has been recognized.
2.20. Employee benefits
Employee benefits are recognized as an expense, unless the cost qualifies to be capitalized as an asset.
1.Short-term obligations
Liabilities for wages and salaries, including non-monetary benefits and annual leave that is expected to be settled wholly
within 12 months after the end of the period in which the employees render the related service are recognized in respect
of employees’ services up to the end of the reporting period. These liabilities are measured at the amounts expected to
be paid when the liabilities are settled. The liabilities are presented as current employee benefit obligations in the
consolidated statement of financial position.
2.Other long-term employee benefit obligations
The liabilities for long service leave are not expected to be settled wholly within 12 months after the end of the period in
which the employees render the related service. They are therefore measured as the present value of expected future
payments to be made in respect of services provided by employees up to the end of the reporting period using the
projected unit credit method. Consideration is given to expected future wage and salary levels, experience of employee
departures and periods of service. Expected future payments are discounted using market yields at the end of the
reporting period of high-quality corporate bonds with terms and currencies that match, as closely as possible, the
estimated future cash outflows. Remeasurements as a result of experience adjustments and changes in actuarial
assumptions are recognized in profit or loss.
The obligations are presented as current liabilities in the consolidated statement of financial position if the entity does
not have an unconditional right to defer settlement for at least 12 months after the reporting period, regardless of when
the actual settlement is expected to occur.
3.Share-based payments
Equity-settled share-based compensation benefits are provided to certain employees. Equity-settled transactions are
awards of shares, options or performance rights over shares, that are provided to employees. The cost of equity-settled
transactions is measured at fair value on grant date. Fair value is determined using the Black-Scholes option pricing
model that takes into account the exercise price, the term of the option, the impact of dilution, the share price at grant
date and expected price volatility of the underlying share, the expected dividend yield and the risk-free interest rate for
the term of the option and volatility. No account is taken of any other vesting conditions.
If the non-vesting condition is within the control of the consolidated entity or employee, the failure to satisfy the
condition is treated as a cancellation. If the condition is not within the control of the consolidated entity or employee and
is not satisfied during the vesting period, any remaining expense for the award is recognized over the remaining vesting
period, unless the award is forfeited. If equity-settled awards are cancelled, it is treated as if it has vested on the date of
cancellation, and any remaining expense is recognized immediately. If a new replacement award is substituted for the
cancelled award, the cancelled and new awards are treated as if they were a modification.
4.Termination benefits
Termination benefits are payable when employment is terminated by the Group before the normal retirement date, or
when an employee accepts voluntary redundancy in exchange for these benefits. The Group recognizes termination
benefits at the earlier of the following dates:
•when the Group can no longer withdraw the offer of those benefits, and
•when the entity recognizes costs for a restructuring that is within the scope of IAS 37 Provisions, Contingent
Liabilities and Contingent Assets and involves the payment of termination benefits. In the case of an offer made to
F-18
encourage voluntary redundancy, the termination benefits are measured based on the number of employees
expected to accept the offer. Benefits falling due more than 12 months after the end of the reporting period are
discounted to present value.
2.21. Borrowings
Borrowings are initially recognized at fair value, net of transaction costs incurred. Borrowings are subsequently measured
at amortized cost. Any difference between the proceeds (net of transaction costs) and the redemption amount is
recognized in profit or loss over the period of the borrowings using the effective interest method. Fees paid on the
establishment of loan facilities are recognized as transaction costs of the loan to the extent that it is probable that some
or all of the facility will be drawn down. In this case, the fee is deferred until the draw- down occurs. To the extent there
is no evidence that it is probable that some or all of the facility will be drawn down, the fee is capitalized as a prepayment
for liquidity services and amortized over the period of the facility to which it relates.
The fair value of the liability portion of a convertible bond is determined using a market interest rate for an equivalent
non-convertible bond. This amount is recorded as a liability on an amortized cost basis until extinguished on conversion
or maturity of the bonds. The remainder of the proceeds is allocated to the conversion option. This is recognized and
included in share capital reserve within equity.
Borrowing costs that are directly attributable to the construction of qualifying assets are capitalized as part of the cost of
the relevant asset.
Borrowings are removed from the consolidated statement of financial position when the obligation specified in the
contract is discharged, cancelled or expired. The difference between the carrying amount of a financial liability that has
been extinguished or transferred to another party and the consideration paid, including any non-cash assets transferred
or liabilities assumed, is recognized in profit or loss as other income or finance costs.
Borrowings are classified as current liabilities unless the Group has an unconditional right to defer settlement of the
liability for at least 12 months after the reporting period.
2.22. Revenue
Revenue is measured at the fair value of the consideration received or receivable. Amounts disclosed as revenue are net
of returns, trade allowances, rebates and amounts collected on behalf of third parties.
Revenue is recognized using a five step approach in accordance with IFRS 15 Revenue from Contracts with Customers to
depict the transfer of promised goods or services to customers in an amount that reflects the consideration to which the
Group expects to be entitled in exchange for those goods or services.
Distinct promises within the contract are identified as performance obligations. The transaction price of the contract is
measured based on the amount of consideration the Group expects to be entitled to from the customer in exchange for
goods or services. Factors such as requirements around variable consideration, significant financing components, non-
cash consideration, or amounts payable to customers also determine the transaction price. The transaction is then
allocated to separate performance obligations in the contract based on relative standalone selling prices.
Revenue is recognized when, or as, performance obligations are satisfied, which is when control of the promised good or
service is transferred to the customer.
Amounts received prior to satisfying the revenue recognition criteria are recorded as contract liabilities. Amounts
expected to be recognized as revenue within the 12 months following the consolidated statement of financial position
date are classified within current liabilities. Amounts not expected to be recognized as revenue within the 12 months
following the consolidated statement of financial position date are classified within non-current liabilities.
1.Sales of goods
The Group’s revenue recognition policy has been updated below to reflect the impact of the acquisition of RLS.
Sales are recognized at a point-in-time when control of the products has transferred, being when the products are
delivered to the customer. Further, in determining whether control has transferred, Telix considers if there is a present
right to payment and legal title, along with risks and rewards of ownership having transferred to the customer. Revenue
from sales is recognized based on the price specified in the contract, net of the estimated volume discounts and rebates.
Accumulated experience is used to estimate and provide for discounts, using the expected value method, and revenue is
recognized to the extent that it is highly probable that a significant reversal will not occur. No element of financing is
deemed present as the sales are made with credit terms ranging from 30 to 45 days, which is consistent with market
practice.
Where the Group uses third-party distributors to facilitate the supply of a product, a fee is charged for the radiolabeling
process, with control transferring to the customer once the radiolabeling process is completed. This fee is expensed
within Cost of sales in the Consolidated statement of comprehensive income or loss.
F-19
With the acquisition of RLS, the Group has the ability to perform the radiolabeling process for existing Telix products. The
cost of radiolabeling for Telix products is recognized within Cost of sales in the Consolidated statement of
comprehensive income or loss.
2.Licenses of intellectual property
When licenses of intellectual property are distinct from other goods or services promised in the contract, the transaction
price is allocated to the license as revenue upon transfer of control of the license to the customer. All other promised
goods or services in the license agreement are evaluated to determine if they are distinct. If they are not distinct, they
are combined with other promised goods or services.
The transaction price allocated to the license performance obligation is recognized based on the nature of the license
arrangement. The transaction price is recognized over time if the nature of the license is a ‘right to access’ license. This is
where the Group performs activities that significantly affect the intellectual property to which the customer has rights,
the rights granted by the license directly expose the customer to any positive or negative effects of the Group’s
activities, and those activities do not result in the transfer of a good or service to the customer as those activities occur.
When licenses do not meet the criteria to be a right to access license, the license is a ’right to use’ license, and the
transaction price is recognized at the point in time when the customer obtains control over the license.
3.Research and development services
Where research and development ("R&D") services do not significantly modify or customize the license nor are the
license and development services significantly interrelated or interdependent, the provision of R&D services is
considered to be distinct. The transaction price is allocated to the R&D services based on a cost-plus margin approach.
Revenue is recognized over time based on the costs incurred to date as a percentage of total forecast costs.
Reforecasting of total costs is performed at the end of each reporting period to ensure that costs recognized represent
the goods or services transferred.
4.Manufacturing services
Revenue from providing contract manufacturing services is recognized in the period in which the services are rendered.
For fixed-price contracts, revenue is recognized based on the actual service provided to the end of the reporting period
as a proportion of the total services to be provided, because the customer receives and uses the benefits
simultaneously. This is determined based on the actual time spent to deliver the service relative to the total expected
hours.
For instances where contracts include multiple deliverables, such as the sale of consumables and irradiation systems,
each deliverable is therefore accounted for as a separate performance obligation. Where the contracts include multiple
performance obligations, the transaction price is allocated to each performance obligation based on the stand-alone
selling prices. Where these are not directly observable, they are estimated based on expected cost plus margin. If
contracts include the installation of systems, revenue for the system is recognized at a point in time when control is
transferred to the customer. The customer obtains control at the point in time when the system is delivered to the
customer in accordance with the agreed terms and the customer accepted the system.
5.Financing component
The existence of a significant financing component in the contract is considered under the five-step method under IFRS
15 Revenue from Contracts with Customers.
If the timing of payments agreed to by the parties to the contract (either explicitly or implicitly) provides the customer or
the Group with a significant benefit of financing the transfer of goods or services to the customer, the promised amount
of consideration will be adjusted for the effects of the time value of money when determining the transaction price.
6.Milestone revenue
The five-step method under IFRS 15 Revenue from Contracts with Customers is applied to measure and recognize
milestone revenue.
The receipt of milestone payments is often contingent on meeting certain clinical, regulatory or commercial targets, and
is therefore considered variable consideration.
The transaction price of the contingent milestone is estimated using the most likely amount method. Within the
transaction price, some or all of the amount of the contingent milestone is included only to the extent that it is highly
probable that a significant reversal in the amount of cumulative revenue recognized will not occur when the uncertainty
associated with the contingent milestone is subsequently resolved. Milestone payments that are not within the control of
the Group, such as regulatory approvals, are not considered highly probable of being achieved until those approvals are
received.
Any changes in the transaction price are allocated to all performance obligations in the contract unless the variable
consideration relates only to one or more, but not all, of the performance obligations. When consideration for milestones
is a sale-based or usage-based royalty that arises from licenses of intellectual property (such as cumulative net sales
F-20
targets), revenue is recognized at the later of when (or as) the subsequent sale or usage occurs, or when the
performance obligation to which some or all of the royalty has been allocated has been satisfied (or partially satisfied).
7.Sales-based or usage-based royalties
Licenses of intellectual property can include royalties that are based on the customer’s usage of the intellectual property
or sale of products that contain the intellectual property. The specific exception to the general requirements of variable
consideration and the constraint on variable consideration for sales-based or usage-based royalties promised in a
license of intellectual property is applied. The exception requires such revenue to be recognized at the later of when (or
as) the subsequent sale or usage occurs and the performance obligation to which some or all of the sales-based or
usage-based royalty has been allocated has been satisfied (or partially satisfied).
8. Distribution services
Where RLS is a distributor for third-party customers, revenue is recognized via a service fee when it satisfies the
performance obligation of the radiolabeling process. On completion of the radiolabeling process, control of the product
transfers to RLS, which also recognizes revenue from the sale of goods once all performance obligations have been
satisfied with the customer. These patient specific doses are transported and delivered to imaging clinics within specific
time frames. Revenue is recognized in Revenue from contracts with customers within the Consolidated statement of
comprehensive income and loss.
Distribution services contracts are generally fixed-price per dose and as such revenue is recognized on successful
delivery of a ready-to-inject dose, as the customer receives and uses the benefits simultaneously.
2.23. Government grants
Income from government grants is recognized at fair value where there is a reasonable assurance that the grant will be
received, and the Group will comply with all attached conditions. Income from government grants is recognized in the
consolidated statement of comprehensive income or loss on a systematic basis over the periods in which the Group
recognizes as an expense the related costs for which the grants are intended to compensate.
2.24. Income tax
The income tax expense or credit for the period is the tax payable on the current period’s taxable income based on the
applicable income tax rate for each jurisdiction adjusted by changes in deferred tax assets and liabilities attributable to
temporary differences and to unused tax losses.
Deferred income tax is provided in full, using the liability method, on temporary differences arising between the tax bases
of assets and liabilities and their carrying amounts in the consolidated financial statements. However, deferred tax
liabilities are not recognized if they arise from the initial recognition of goodwill. Deferred income tax is also not
accounted for if it arises from initial recognition of an asset or liability in a transaction other than a business combination
that at the time of the transaction affects neither accounting nor taxable profit or loss. Deferred income tax is determined
using tax rates (and laws) that have been enacted or substantively enacted by the end of the reporting period and are
expected to apply when the related deferred income tax asset is realized or the deferred income tax liability is settled.
Deferred tax assets are recognized only if it is probable that future taxable amounts will be available to utilize those
temporary differences and losses.
Included in income tax expense for the period is the effect of Australian R&D tax credits which may only be offset against
Australian taxable income. As such, they are recognized as a component of income tax expense.
Tax consolidation regime
Telix Pharmaceuticals Limited and its wholly owned Australian resident entities have formed a tax-consolidated group
and are therefore taxed as a single entity. The head entity within the tax-consolidated group is Telix Pharmaceuticals
Limited. As a consequence, the deferred tax assets and deferred tax liabilities of these entities have been offset in the
consolidated financial statements.
2.25. Sales taxes and Goods and Services Tax (GST)
Revenues, expenses and assets are recognized net of the amount of associated sales taxes and GST, unless the GST
incurred is not recoverable from the taxation authority. In this case it is recognized as part of the cost of acquisition of
the asset or as part of the expense.
Cash flows are presented on a gross basis. The GST components of cash flows arising from investing or financing
activities which are recoverable from, or payable to the taxation authority, are presented as operating cash flows.
2.26. Earnings per share
1.Basic earnings per share
Basic earnings per share is calculated by dividing: the profit attributable to owners of the Company, excluding any costs
of servicing equity other than ordinary shares, by the weighted average number of ordinary shares outstanding during
F-21
the financial period, adjusted for bonus elements in ordinary shares issued during the period and excluding treasury
shares.
2.Diluted earnings per share
Diluted earnings per share adjusts the figures used in the determination of basic earnings per share to take into account:
the after-income tax effect of interest and other financing costs associated with dilutive potential ordinary shares, and
the weighted average number of additional ordinary shares that would have been outstanding assuming the conversion
of all dilutive potential ordinary shares.
2.27. Fair value measurement
Certain judgments and estimates are made in determining the fair values of the financial instruments that are recognized
and measured at fair value in the financial statements. To provide an indication about the reliability of the inputs used in
determining fair value, the Group has classified its financial instruments into the three levels prescribed under the
accounting standards. The different levels have been defined as follows:
•Level 1: fair value of financial instruments traded in active markets is based on quoted market prices at the end of
the reporting period. The quoted market price used for financial assets is the current bid price.
•Level 2: fair value of financial instruments that are not traded in an active market is determined using valuation
techniques which maximize the use of observable market data and rely as little as possible on entity specific
estimates. If all significant inputs required to fair value an instrument are observable, the instrument is included in
level 2.
•Level 3: if one or more of the significant inputs is not based on observable market data, the instrument is included in
level 3.
There were no transfers between level 1, 2 and 3 for recurring fair value measurements during the year. The Group’s
policy is to recognize transfers into and transfers out of fair value hierarchy levels at the end of the reporting period.
Certain judgments and estimates are made in determining the fair values of the financial instruments that are recognized
and measured at fair value in the financial statements.
2.28. Key judgments and estimates
In the process of applying the Group’s accounting policies, a number of judgments and estimates of future events are
required.
1.Impairment assessment – carrying value of goodwill and intangible assets
The assessment of impairment of the goodwill and intangible assets has required estimates and judgments to be made.
The inputs for these have been outlined in note 21.
2.Contingent consideration and decommissioning liabilities
The Group has identified the contingent consideration and decommissioning liabilities as balances requiring estimates
and significant judgments. These estimates and judgments have been outlined in note 27 and note 28.
3. Segment reporting
The Group has operations in the Americas, Asia Pacific, and Europe, Middle East and Africa regions.
Reportable segments
The Group’s operating segments are based on the reports reviewed by the Group Chief Executive Officer who is
considered to be the chief operating decision maker.
Segment performance is evaluated based on Adjusted earnings before interest, tax, depreciation and amortization
("Adjusted EBITDA"). Adjusted EBITDA excludes the effects of the remeasurement of contingent consideration and
government grant liabilities and other income and expenses which may have an impact on the quality of earnings such as
impairments where the impairment is the result of an isolated, non-recurring event. Interest income and treasury related
finance costs are not allocated to segments as this activity is managed by a central treasury function, which manages
the cash position of the Group.
F-22
Segment assets and liabilities are measured in the same way as in the financial statements. The assets and liabilities are
allocated based on the operations of the segment.
Reportable segment | Principal activities |
Precision Medicine | Commercial sales of Illuccix, Gozellix and other diagnostic products subsequent to obtaining regulatory approvals. This segment includes the development activities of the Group’s diagnostic pipeline. The Group’s International and Medical Technologies businesses are operating segments that are included within the Precision Medicine reportable segment due to the similar nature of the diagnostic products being sold or developed for commercialization. |
Therapeutics | Developing the Group’s core therapeutic pipeline for commercialization. This segment includes revenue received from license agreements prior to commercialization and research and development services. This segment includes the development activities of the Group’s therapeutic pipeline. |
Manufacturing Solutions | Telix Manufacturing Solutions business. This segment comprises costs to operate our facilities and assets associated with the Group’s vertically integrated manufacturing and supply chain. This business includes facilities at Brussels South, IsoTherapeutics, TMS Sacramento, North Melbourne, ARTMS and RLS Radiopharmacies. |
Reconciling items includes head office and centrally managed costs.
Precision Medicine | Therapeutics | Manufacturing Solutions | Inter- segment eliminations | Total segment | 1. | |||||
2025 | US$'000 | US$'000 | US$'000 | US$'000 | US$'000 | |||||
Revenue from contracts with customers | ||||||||||
Inter-segment revenue | ( | |||||||||
Cost of sales | ( | ( | ( | ( | ||||||
Gross profit | ( | |||||||||
Research and development costs | ( | ( | ( | ( | ||||||
Selling and marketing expenses | ( | ( | ( | ( | ||||||
Manufacturing and distribution costs | ( | ( | ( | ( | ||||||
General and administration costs | ( | ( | ( | ( | ||||||
Other (losses)/gains (net) | ( | |||||||||
Operating profit | ( | ( | ( | |||||||
Other losses/(gains) (net) | ( | ( | ( | |||||||
Depreciation and amortization | ||||||||||
Adjusted earnings/ (loss) before interest, tax, depreciation and amortization | ( | ( | ( |
F-23
Precision Medicine | Therapeutics | Manufacturing Solutions | Total segment | 1. | ||||
US$'000 | US$'000 | US$'000 | US$'000 | |||||
2024 | (Recast) | (Recast) | (Recast) | (Recast) | ||||
Revenue from contracts with customers | ||||||||
Cost of sales | ( | ( | ( | |||||
Gross profit | ( | |||||||
Research and development costs | ( | ( | ( | ( | ||||
Selling and marketing expenses | ( | ( | ( | ( | ||||
Manufacturing and distribution costs | ( | ( | ( | ( | ||||
General and administration costs | ( | ( | ( | ( | ||||
Other (losses)/gains (net) | ( | ( | ||||||
Operating profit/(loss) | ( | ( | ||||||
Other losses/(gains) (net) | ( | |||||||
Depreciation and amortization | ||||||||
Adjusted earnings/(loss) before interest, tax, depreciation and amortization | ( | ( |
Precision Medicine | Therapeutics | Manufacturing Solutions | Total segment | |||||
US$'000 | US$'000 | US$'000 | US$'000 | |||||
2023 | (Recast) | (Recast) | (Recast) | (Recast) | ||||
Revenue from contracts with customers | ||||||||
Cost of sales | ( | ( | ||||||
Gross profit | ||||||||
Research and development costs | ( | ( | ( | ( | ||||
Selling and marketing expenses | ( | ( | ( | |||||
Manufacturing and distribution costs | ( | ( | ( | ( | ||||
General and administration costs | ( | ( | ( | ( | ||||
Other losses (net) | ( | ( | ||||||
Operating profit/(loss) | ( | ( | ||||||
Other losses (net) | ||||||||
Depreciation and amortization | ||||||||
Adjusted earnings/(loss) before interest, tax, depreciation and amortization | ( | ( |
Activities between segments are eliminated on consolidation. The amounts presented are measured consistently with the
group’s external reporting. Segment assets are allocated based on the operations of the segment and the physical
location of the asset.
F-24
2025 | 2024 | 2023 | ||||||
Note | US$'000 | US$'000 | US$'000 | |||||
(Recast) | (Recast) | |||||||
Total segment adjusted EBITDA | ||||||||
Unallocated income, expenses and eliminations: | ||||||||
General and administration costs | ( | ( | ( | |||||
Other gains/(losses) (net) | 10 | ( | ||||||
Finance income | ||||||||
Finance costs | 11 | ( | ( | ( | ||||
Depreciation and amortization | 9 | ( | ( | ( | ||||
(Loss)/profit before income tax | ( |
General and administration costs predominantly comprise employment costs of $31,950,000 (2024: $30,264,000 2023:
$18,420,000 ) and other centrally managed IT, legal and other corporate costs.
Precision Medicine | Therapeutics | Manufacturing Solutions | Total segment | Reconciling items | Group | 1. | ||||||
December 31, 2025 | US$'000 | US$'000 | US$'000 | US$'000 | US$'000 | US$'000 | ||||||
Total assets | ||||||||||||
Total liabilities | ||||||||||||
Additions to non- current assets |
Precision Medicine | Therapeutics | Manufacturing Solutions | Total segment | Reconciling items | Group | |||||||
US$'000 | US$'000 | US$'000 | US$'000 | US$'000 | US$'000 | |||||||
December 31, 2024 | (Recast) | (Recast) | (Recast) | (Recast) | (Recast) | (Recast) | ||||||
Total assets | ||||||||||||
Total liabilities | ||||||||||||
Additions to non- current assets |
Reconciling items primarily comprise cash and cash equivalents held centrally $82,833,000 (2024: $326,421,000 ),
investments in financial assets $37,094,000 (2024: $34,746,000 ), property, plant and equipment $6,915,000 (2024:
$1,084,000 ), borrowings (convertible bonds) $383,023,000 (2024: $333,378,000 ) which are managed centrally.
F-25
2025 | 2024 | 2023 | |||
Revenue by location of customer | Revenue by location of customer | Revenue by location of customer | |||
US$'000 | US$'000 | US$'000 | |||
(Recast) | (Recast) | ||||
China | |||||
United States | |||||
Other countries | |||||
Total |
2025 | 2025 | 2024 | |
Non-current assets by location of asset | Non-current assets by location of asset | ||
US$'000 | US$'000 | ||
(Recast) | |||
Australia | |||
Belgium | |||
Canada | |||
United Kingdom | |||
United States | |||
Other countries | |||
Total |
The total non-current assets figure above excludes deferred tax assets.
F-26
4. Revenue from contracts with customers
Disaggregation of revenue from contracts with customers.
following major business activities:
2025 | 2024 | 2023 | ||||||
Recognition | Operating segment | US$'000 | US$'000 | US$'000 | ||||
(Recast) | (Recast) | |||||||
Sale of goods | At a point in time | Precision Medicine | ||||||
Sale of goods | At a point in time | Manufacturing Solutions | ||||||
Royalty income | At a point in time | Precision Medicine | ||||||
Royalty income | At a point in time | Therapeutics | ||||||
Provision of services | Over time | Manufacturing Solutions | ||||||
Licenses of intellectual property | Over time | Therapeutics | ||||||
Research and development services | Over time | Precision Medicine | ||||||
Research and development services | Over time | Therapeutics | ||||||
Total revenue from continuing operations |
5. Research and development costs
2025 | 2024 | 2023 | ||||
US$'000 | US$'000 | US$'000 | ||||
Therapeutics | (Recast) | (Recast) | 1. | |||
Phase 3 | ||||||
Phase 2 | ||||||
Early stage clinical candidates | ||||||
Pre-clinical research and innovation | ||||||
Total Therapeutics R&D | ||||||
Precision Medicine | ||||||
Commercial | ||||||
Pre-commercial1 | ||||||
Pre-clinical research and innovation | ||||||
Total Precision Medicine R&D | ||||||
Total product development R&D | ||||||
Manufacturing Solutions | ||||||
Other research and development projects | ||||||
Total Manufacturing Solutions R&D | ||||||
Inter-segment R&D | ( | |||||
Total research and development costs |
F-27
Other research and development projects includes research and innovation costs and other early-stage development
projects
6. Manufacturing and distribution costs
2025 | 2024 | 2023 | |||
US$'000 | US$'000 | US$'000 | |||
(Recast) | (Recast) | ||||
Radiopharmacy operations | |||||
Quality costs | |||||
Supply chain costs | |||||
Technical services | |||||
Global manufacturing costs |
The increase in manufacturing and distribution costs was driven by the impact of RLS results for 11 months and higher
7. General and administration costs
2025 | 2024 | 2023 | ||||
US$'000 | US$'000 | US$'000 | ||||
(Recast) | (Recast) | |||||
Professional fees | ||||||
Acquisition transaction costs | ||||||
U.S. listing costs | ||||||
IT infrastructure, hosting and support | ||||||
Travel, conferences and entertainment | ||||||
Rent and insurance |
The increase in general and administration costs was driven by the impact of RLS results for 11 months, costs incurred to
implement a Sarbanes-Oxley program, and professional fees in connection with the legal matters outlined in note 34 of
this report. Other material general and administration costs are employment related as included within note 8 below.
8. Employment costs
2025 | 2024 | 2023 | ||||
US$'000 | US$'000 | US$'000 | 1. | |||
(Recast) | (Recast) | |||||
Salaries and wages | ||||||
Short term incentives | ||||||
Sales commissions | ||||||
Share-based payment charge | ||||||
Superannuation | ||||||
Non-Executive Directors’ fees | ||||||
1 Refer to note 28 for further information of remeasurements associated with contingent consideration.
F-28
Salaries and wages of $2,235,000 (2024: $3,984,000 , 2023: $983,000 ) are included within the cost of sales in the
Consolidated statement of comprehensive income/(loss). The increase in employment costs was predominantly due to
the impact of RLS results for 11 months, and additional employees hired to drive higher commercial sales and support our
global manufacturing facilities.
9. Depreciation and amortization
2025 | 2024 | 2023 | ||||
US$'000 | US$'000 | US$'000 | ||||
(Recast) | (Recast) | |||||
Amortization of intangible assets | ||||||
Depreciation | ||||||
10. Other (gains)/losses (net)
2025 | 2024 | 2023 | ||||
US$'000 | US$'000 | US$'000 | ||||
(Recast) | (Recast) | |||||
Remeasurement of contingent consideration1 | ( | |||||
Remeasurement of provisions | ( | ( | ||||
Realized currency gain | ( | ( | ( | |||
Impairments/(impairment reversals) of intangible assets | ( | |||||
Other income | ( | ( | ( | |||
Unrealized currency gain/(loss) | ( | |||||
( | ( |
11. Finance costs
2025 | 2024 | 2023 | ||||
US$'000 | US$'000 | US$'000 | ||||
(Recast) | (Recast) | |||||
Unwind of discount | ||||||
Interest expense on lease liabilities | ||||||
Convertible bond interest expense | ||||||
Interest expense | ||||||
Bank fees | ||||||
Finance costs |
The Group recognized an unwind of discount on convertible bonds of $22,502,000 (2024: $9,088,000 , 2023: Nil ),
unwind of working capital facility costs of $49,000 (2024: Nil , 2023: Nil ), contingent consideration liabilities of
$2,201,000 (2024: $9,546,000 , 2023: $7,575,000 ), provisions of $445,000 (2024: $256,000 , 2023: $279,000 ) and
contract liabilities of $137,000 (2024: $469,000 , 2023: $644,000 ).
F-29
12. Income tax expense/(benefit)
2025 | 2024 | 2023 | ||||
US$'000 | US$'000 | US$'000 | ||||
(Recast) | (Recast) | |||||
Current tax expense1 | ||||||
Deferred tax benefit | ( | ( | ( | |||
Income tax expense/(benefit) | ( |
1.The current tax expense is attributable to Telix Innovations SA and Telix Pharmaceuticals US Inc and is driven by the
individual entity’s taxable profits.
2025 | 2024 | 2023 | ||||
US$'000 | US$'000 | US$'000 | ||||
(Recast) | (Recast) | |||||
(Loss)/profit before income tax | ( | |||||
Prima-facie tax at a rate of | ( | |||||
Tax effect of amounts which are not deductible (taxable) in calculating taxable income: | ||||||
Net R&D tax incentive credit | ( | ( | ( | |||
Remeasurement of provisions | ||||||
Share-based payments expense & Employee Share Trust payments | ( | ( | ( | |||
Sundry items | ||||||
Foreign exchange translation gains | ||||||
( | ||||||
Current year tax losses not recognized | ||||||
Difference in overseas tax rates | ( | ( | ( | |||
Income tax expense/(benefit) | ( |
13. Earnings per share
13.1. Basic earnings per share
2025 | 2024 | 2023 | ||||
Cents | Cents | Cents | 1. | |||
(Recast) | (Recast) | 1. | ||||
Basic (loss)/earnings per share from continuing operations attributable to the ordinary equity holders of the Company | ( | |||||
Total basic (loss)/earnings per share attributable to the ordinary equity holders of the Company | ( |
F-30
2025 | 2024 | 2023 | ||||
Cents | Cents | Cents | 1. | |||
(Recast) | (Recast) | 1. | ||||
Diluted (loss)/earnings per share from continuing operations attributable to the ordinary equity holders of the Company | ( | |||||
Total diluted (loss)/earnings per share attributable to the ordinary equity holders of the Company | ( |
2025 | 2024 | 2023 | ||||
Number | Number | Number | 1. | |||
’000 | ’000 | ’000 | ||||
Weighted average number of ordinary shares used as the denominator in calculating basic earnings/loss per share | ||||||
Weighted average number of ordinary shares used as the denominator in calculating diluted earnings/loss per share1 |
13.3.1.Options and rights
Equity instruments (options, PSARs, PSIRs and rights) granted to employees under the Group’s EIP scheme (refer to note
31 for further details) and rights issued as part of acquisitions are considered to be potential ordinary shares. They have
been included in the determination of diluted earnings per share based on achieving the required performance hurdles,
and to the extent to which they are dilutive.
13.3.2.Convertible bonds
Convertible bonds issued during the year are not included in the calculation of diluted earnings per share, because they
are antidilutive for the year ended December 31, 2025. These options could potentially dilute basic earnings per share in
the future. Refer to note 25.3 for further details relating to the convertible bonds.
14. Trade and other receivables
2025 | 2024 | |||
US$'000 | US$'000 | |||
(Recast) | ||||
Trade receivables | ||||
Allowance for impairment losses2 | ( | ( | ||
F-31
15. Inventories
2025 | 2024 | |||
US$'000 | US$'000 | |||
(Recast) | ||||
Raw materials and stores | ||||
Work in progress | ||||
Finished goods | ||||
Provision for obsolescence | ( | ( | ||
Total inventories |
The amount of inventory recognized as an expense during the year was $142,002,000 (2024: $23,875,000 , 2023:
$13,939,000 ).
Clinical inventory and inventory manufactured as part of the TLX250-Px (Zircaix)1 commercial manufacturing process
qualification and validation has been capitalized as work in progress, with a corresponding provision for obsolescence
recognized. This is on the basis that, prior to regulatory approval, the Group has not demonstrated that the batches
produced can be sold commercially.
16. Other current assets
2025 | 2024 | |||
US$'000 | US$'000 | |||
(Recast) | ||||
Other receivables | ||||
GST receivables | ||||
Prepayments | ||||
Total other current assets |
17. Financial assets
2025 | 2024 | |||
US$'000 | US$'000 | |||
(Recast) | ||||
Investment in Mauna Kea Technologies | ||||
Investment in Atonco SAS | ||||
Investment in IRMA Surgical Pty Ltd | ||||
Restricted cash2 | ||||
Total financial assets |
F-32
18. Deferred tax assets and liabilities
18.1. Deferred tax assets
2025 | 2024 | |||
US$'000 | US$'000 | |||
(Recast) | ||||
The balance comprises temporary differences attributable to: | ||||
Tax losses | ||||
Intangible assets | ||||
Employee benefit obligations | ||||
Lease liabilities | ||||
Inventories | ||||
Other | ||||
Total deferred tax assets | ||||
Set-off of deferred tax liabilities pursuant to set-off provisions | ( | ( | ||
Net deferred tax assets |
Tax losses | Intangible assets | Employee benefit obligations | Lease liabilities | Inventories | Other | Total | 1. | |||||||
Deferred tax assets movements | US$'000 | US$'000 | US$'000 | US$'000 | US$'000 | US$'000 | US$'000 | 1. | ||||||
Balance at January 1, 2025 | ||||||||||||||
Credited/(charged): | ||||||||||||||
on acquisition | ||||||||||||||
to profit and loss | ( | ( | ( | |||||||||||
Balance at December 31, 2025 | ||||||||||||||
(Recast) | (Recast) | (Recast) | (Recast) | (Recast) | (Recast) | (Recast) | ||||||||
Balance at January 1, 2024 | ||||||||||||||
Credited/(charged): | - | |||||||||||||
to profit and loss | ( | |||||||||||||
Balance at December 31, 2024 |
F-33
18.2. Deferred tax liabilities
2025 | 2024 | |||
US$'000 | US$'000 | |||
(Recast) | ||||
The balance comprises temporary differences attributable to: | ||||
Intangible assets | ||||
Right-of-use assets | ||||
Unrealized foreign exchange gains | ||||
Total deferred tax liabilities | ||||
Set-off of deferred tax assets pursuant to set-off provisions | ( | ( | ||
Net deferred tax liabilities |
Intangible assets | Right-of- use assets | Unrealized foreign exchange gains | Total | |||||
Deferred tax liabilities movements | US$'000 | US$'000 | US$'000 | US$'000 | ||||
Balance at January 1, 2025 | ||||||||
Charged/(credited): | ||||||||
on acquisition | ||||||||
to profit and loss | ( | |||||||
Balance at December 31, 2025 | ||||||||
(Recast) | (Recast) | (Recast) | (Recast) | |||||
Balance at January 1, 2024 | ||||||||
Charged/(credited): | ||||||||
on acquisition | ||||||||
to profit and loss | ( | |||||||
Balance at December 31, 2024 |
18.3. Unrecognized deferred tax assets
2025 | 2024 | |||
Unrecognized deferred tax assets | US$'000 | US$'000 | ||
(Recast) | ||||
Tax losses and tax credits | ||||
Temporary differences in relation to provisions | ||||
Temporary differences in relation to employee benefit obligations | ||||
Temporary differences in relation to intangible assets | ||||
Temporary differences in relation to inventories | ||||
Temporary differences in relation to lease liabilities | ||||
Temporary differences in relation to share-based payments | ||||
Total unrecognized deferred tax assets |
F-34
18.4. Carried forward tax losses and credits
Unused tax losses and carried forward tax credits for which no deferred tax asset has been recognized | 2025 | 2024 | 2023 | |||
US$'000 | US$'000 | US$'000 | ||||
(Recast) | (Recast) | |||||
Australia | ||||||
Other countries | ||||||
Unrecognized income tax benefit | ||||||
Unused tax losses and carried forward tax credits for which a deferred tax asset has been recognized | ||||||
Other countries | ||||||
Recognized income tax benefit |
19. Property, plant and equipment
Land and buildings | Plant, equipment and vehicles | Furniture, fittings and equipment | Leasehold improvements | Construction in progress | Total | 1. | ||||||
US$'000 | US$'000 | US$'000 | US$'000 | US$'000 | US$'000 | |||||||
Balance at January 1, 2025 | ||||||||||||
Additions | ||||||||||||
Acquisition of businesses | ||||||||||||
Reclassifications | ( | ( | ( | |||||||||
Changes in provisions | ( | ( | ( | |||||||||
Depreciation charge | ( | ( | ( | ( | ( | |||||||
Exchange differences | ||||||||||||
Balance at December 31, 2025 | ||||||||||||
Cost | ||||||||||||
Accumulated depreciation | ( | ( | ( | ( | ( | |||||||
Net book amount | ||||||||||||
(Recast) | (Recast) | (Recast) | (Recast) | (Recast) | (Recast) | |||||||
Balance as at January 1, 2024 | ||||||||||||
Additions | ||||||||||||
Acquisition of businesses | ||||||||||||
Reclassifications | ( | ( | ||||||||||
Changes in provisions | ||||||||||||
Depreciation charge | ( | ( | ( | ( | ||||||||
Exchange differences | ( | ( | ( | ( | ( | |||||||
Balance at December 31, 2024 | ||||||||||||
Cost | ||||||||||||
Accumulated depreciation | ( | ( | ( | ( | ( | |||||||
Net book amount |
F-35
20. Right-of-use assets
Properties | Motor vehicles | Total | 1. | |||
US$'000 | US$'000 | US$'000 | ||||
Balance at January 1, 2025 | ||||||
Additions | ||||||
Acquisition of businesses | ||||||
Depreciation charge | ( | ( | ( | |||
Exchange differences | ||||||
Balance at December 31, 2025 | ||||||
Cost | ||||||
Accumulated depreciation | ( | ( | ( | |||
Net book amount | ||||||
(Recast) | (Recast) | (Recast) | ||||
Balance at January 1, 2024 | ||||||
Additions | ||||||
Reclassifications | ||||||
Depreciation charge | ( | ( | ( | |||
Exchange differences | ( | ( | ( | |||
Balance at December 31, 2024 | ||||||
Cost | ||||||
Accumulated depreciation | ( | ( | ( | |||
Net book amount |
assets:
2025 | 2024 | 2023 | ||||
Depreciation charge on right-of-use assets | US$'000 | US$'000 | US$'000 | |||
(Recast) | (Recast) | |||||
Properties | ||||||
Motor vehicles | ||||||
F-36
21. Intangible assets
Goodwill | Intellectual property | Customer relationships and brands | Software | Patents | Licenses | Total | 1. | |||||||
US$'000 | US$'000 | US$'000 | US$'000 | US$'000 | US$'000 | US$'000 | 1. | |||||||
Balance at January 1, 2025 | ||||||||||||||
Acquisition of businesses | ||||||||||||||
Additions | - | |||||||||||||
Measurement period adjustments | ||||||||||||||
Reclassifications | ( | |||||||||||||
Amortization charge | - | ( | ( | ( | ( | ( | ( | |||||||
Impairments | - | ( | ( | |||||||||||
Changes in provisions | ( | ( | ||||||||||||
Exchange differences | ||||||||||||||
Balance at December 31, 2025 | ||||||||||||||
Cost | ||||||||||||||
Accumulated amortization | ( | ( | ( | ( | ( | ( | ||||||||
Net book amount | ||||||||||||||
(Recast) | (Recast) | (Recast) | (Recast) | (Recast) | (Recast) | (Recast) | ||||||||
Balance as at January 1, 2024 | ||||||||||||||
Acquisition of businesses | ||||||||||||||
Additions | - | |||||||||||||
Reclassifications | ( | |||||||||||||
Amortization charge | - | ( | ( | ( | ( | ( | ||||||||
Impairments | - | |||||||||||||
Changes in provisions | ||||||||||||||
Exchange differences | ( | ( | ( | ( | ( | ( | ( | |||||||
Balance at December 31, 2024 | ||||||||||||||
Cost | ||||||||||||||
Accumulated amortization | ( | ( | ( | ( | ( | |||||||||
Net book amount |
F-37
Cash generating units
2025 | 2024 | |||||||
Operating segment | Useful life | Product or business unit | US$'000 | US$'000 | 1. | |||
(Recast) | 1. | |||||||
Precision Medicine | Definite and indefinite | TLX591-Px (Illuccix®) | ||||||
Precision Medicine | Definite | TLX66-Px | ||||||
Precision Medicine | Indefinite | TLX300-Px | ||||||
Precision Medicine | Definite | Patents | ||||||
Precision Medicine | Definite and indefinite | SENSEI | ||||||
Precision Medicine | Indefinite | Dedicaid, QDOSE | ||||||
Therapeutics | Indefinite | TLX101-Tx | ||||||
Therapeutics | Indefinite | TLX090-Tx | ||||||
Therapeutics | Indefinite | TLX591-Tx | ||||||
Therapeutics | Indefinite | TLX66-Tx | ||||||
Therapeutics | Indefinite | TLX300-Tx | ||||||
Therapeutics | Indefinite | TLX400-Tx | ||||||
Therapeutics | Indefinite | Telix Targeting Technology | ||||||
Manufacturing Solutions | Indefinite | ARTMS | ||||||
Manufacturing Solutions | Indefinite | RLS | ||||||
Manufacturing Solutions | Definite | RLS | ||||||
Manufacturing Solutions | Definite and indefinite | IsoTherapeutics | ||||||
Manufacturing Solutions | Definite | Brussels South and Sacramento | ||||||
2025 | 2024 | ||
US$'000 | US$'000 | ||
CGU | 1. | (Recast) | |
Brussels South and Sacramento | |||
RLS | |||
IsoTherapeutics | |||
ARTMS | |||
TLX101-Tx | |||
TLX591-Px (Illuccix®) | |||
Total goodwill allocated to CGUs |
For the purposes of the annual impairment test outlined below, each RLS radiopharmacy is considered a separate CGU
on the basis that each site incurs site-specific costs and generates independent cash flows.
Impairment test for goodwill and indefinite life intangible assets
Goodwill and indefinite life intangible assets are tested annually for impairment or more frequently if events or changes in
circumstances indicate that the carrying value of cash-generating units containing indefinite-lived intangible assets and
goodwill may be impaired. Potential impairment is identified by comparing the fair value of a cash-generating unit to its
carrying value, including goodwill. At December 31, 2025, the Directors used a fair value less costs to sell approach to
assess the carrying value of goodwill and indefinite life intangible assets. No impairment was recognized by the Group.
F-38
Key assumptions used for the fair value less costs to sell approach
The Group has identified the estimate of the recoverable amount as a significant judgment for the year ended
December 31, 2025. In determining the recoverable amount of goodwill and indefinite life intangible assets, the Group
has used discounted cash flow forecasts and the following key assumptions (classified as level 3 inputs in the fair value
hierarchy):
•discounted expected future cash flows for assets under development comprise of remaining costs to be incurred to
marketing authorization and then span a further 10 years from marketing authorization. A terminal value with a
declining growth rate, where appropriate, based on our view of the longer term growth profile of the asset is
applied. This reflects the anticipated product life cycle, and includes cash inflows and outflows determined using
further assumptions below
•discounted expected future cash flows for operating business within Manufacturing Solutions span 10 years after
which a terminal value using a long term growth rate of 5 % (2024: 5 %) is applied, and cash inflows and outflows
determined using further assumptions below
•asset specific risk adjusted post-tax discount rates which range from – 11.7 % to 15.0 % (2024: 12.5 %)
•regulatory/marketing authorization approval dates, these are re-assessed in conjunction with Senior Management
and Commercial teams
•expected sales volumes, these are determined by applying a target market share to cancer incidence rates across
various countries, sourced from data provided by the World Health Organization’s International Agency for Research
on Cancer
•net sales price per unit, for commercialized products forecast average selling price is used and for products in
development a target sales price is used
•approval for marketing authorization probability success factor, this varies depending on the clinical trial stage of
each program and is generally based on internal or external clinical research or publically available industry data
•in relation to cash outflows consideration has been given to cost of sales, selling and marketing expenses, general
and administration costs and the anticipated research and development costs to reach commercialization.
Associated expenses such as royalties, milestone payments and license fees are included, and
•costs of disposal were assumed to be immaterial at December 31, 2025.
Impact of possible changes in key assumptions
The Group has considered reasonable possible changes in the key assumptions as outlined below and has not identified
any instances that could cause the carrying amounts of the intangible assets at December 31, 2025 to exceed their
recoverable amounts.
group would have to recognize an impairment against the carrying value of the intangible assets of each respective CGU.
Asset | An increase of 2.5% in post- tax discount rate | A 10% decrease in probability of success | A one year delay in commercialization | A 50% reduction in terminal growth rate | A 10% reduction in future cash inflows |
US$'000 | US$'000 | US$'000 | US$'000 | US$'000 | |
Telix Targeting Technology | ( | ( | ( | Not applicable | Not applicable |
RLS | ( | Not applicable | Not applicable | ( | ( |
The key sensitivities in the valuation remain the continued successful development and commercialization of core assets.
If the Group is unable to successfully develop each asset, this may result in an impairment of the carrying amount of our
intangible assets.
There were no other internal or external factors identified that could result in an impairment of definite life intangible
F-39
22. Other non-current assets
2025 | 2024 | ||
US$'000 | US$'000 | ||
(Recast) | |||
Isotope raw materials | |||
Investment in sub-leases | |||
Other deposits and assets | |||
Total other non-current assets |
23. Acquisitions
23.1 Acquisition of RLS (USA) Inc.
On January 28, 2025 Telix completed the acquisition of RLS (USA) Inc. (RLS), a radiopharmacy network distributing PET,
SPECT and therapeutic radiopharmaceuticals. The acquisition of RLS is aligned to Telix’s investment strategy around
vertically integrated supply chain, manufacturing, and distribution, further enabling the delivery of future clinical and
commercial radiopharmaceutical products.
The total consideration is $240,921,000 of which $225,171,000 was paid in cash. A further $15,750,000 is payable in
cash, contingent on achievement of certain milestones related to demonstration of accretive financial and operational
performance during the four-quarters following closing.
F-40
the acquisition date.
Fair value | |||
Consideration | US$'000 | 1. | |
Cash paid | |||
Contingent consideration | |||
Total consideration | |||
Recognized amounts of identifiable assets acquired and liabilities assumed | |||
Cash and cash equivalents | |||
Trade and other receivables | |||
Other current assets | |||
Inventories | |||
Property, plant and equipment | |||
Intangible assets | |||
Investment in sub-lease | |||
Right-of-use assets | |||
Trade and other payables | ( | ||
Contingent or deferred consideration | ( | ||
Lease liabilities | ( | ||
Total identifiable assets and liabilities | |||
Fair value adjustments | |||
Customer relationships | |||
Brand name | |||
Radiopharmacy licenses | |||
Deferred tax liabilities | ( | ||
Deferred tax asset | |||
Total fair value adjustments | |||
Goodwill | |||
Total |
Following the finalization of the purchase price accounting, goodwill increased by $13,173,000 from the provisional fair
value reported as at June 30, 2025 for the reasons below:
•post acquisition net working capital changes of $2,816,000 , and
•reduction of deferred tax asset by $10,357,000 following the completion of the formal tax analysis assessing the
recoverability of the acquired carried forward tax tax losses.
The goodwill arising is attributable to the acquired workforce, anticipated future cost savings from utilizing RLS'
radiopharmaceutical distribution capabilities and synergies of integrating the business within the Group. The goodwill
arising from the acquisition has been allocated to each radiopharmacy which operate as separate CGUs (refer to note 21
for further details).
Fair value adjustments have been recognized for acquisition-related intangible assets and related deferred tax.
F-41
Acquired intangible asset | Useful economic life |
Customer relationships | |
Brand name | |
Licenses |
Significant estimates used in purchase price accounting
The Group has used the multi-period excess earnings method and the relief from royalty method, which are a form of the
income approach, in estimating the fair value of customer relationships and the brand name acquired, respectively when
allocating the purchase consideration paid for the acquisition.
The estimates of the customer relationships and the brand name involve significant judgment by management in making
cash flow projections for the intangible assets acquired and include significant assumptions with measurement
uncertainty, such as:
•the revenue growth rates, earnings before interest and taxes ("EBIT") margin, and discount rate for customer
relationships, and
•revenue growth rates, royalty rate, and discount rate for the brand name.
RLS contributed $170,088,000 in revenue and a net loss of $8,767,000 towards the Group’s loss before tax attributable
to equity holders of the parent for the period after the date of acquisition.
Had the acquisition of RLS been completed on the first day of the 2025 financial year, Group revenues would have been
approximately $13,607,000 higher and Group loss before tax attributable to equity holders of the parent would have
been approximately $1,484,000 higher.
23.2 Acquisition of ImaginAb, Inc.
On January 30, 2025 Telix completed the acquisition of a pipeline of next generation therapeutic candidates, proprietary
novel biologics technology platform, and a protein engineering and discovery research facility to enhance existing
innovation capabilities from antibody engineering company ImaginAb, Inc.
The upfront purchase price for the transaction was $67,116,000 which comprised $10,000,000 in cash and 29,895,000 in
equity through the issue of 2,053,311 fully paid ordinary Telix shares in January 2025 at a share price of $14.56 per
share. A further $60,000,000 in Milestone Rights, or performance rights, is payable in cash and/or in ordinary shares,
upon achievement of certain clinical milestones. The purchase price also includes a deferred amount payable of
$3,472,000 (up to a maximum of $4,000,000 in equity) at the conclusion of a 15-month indemnity period.
Upon achievement of specific key development and commercial milestones, Telix will pay up to a total of $185 million, a
portion of which may be paid in cash or equity at Telix’s election. Royalties are also payable on net sales in the low single
digits on a limited number of platform and early-stage products after the first four products have been developed, as
well as single-digit sublicense fees, as applicable. The acquisition has been allocated to the Therapeutics operating
segment.
The Group has assessed that the acquisition does not meet the definition of a business in IFRS 3/AASB 3 Business
Combinations as the Group concluded that the acquisition does not include a self-sustaining substantive process
capable of generating outputs. The assets acquired are discovery stage assets and the process of transforming them
into commercially available products remains undeveloped and incomplete at the acquisition date. On this basis, the
Group has concluded that the components acquired will be treated as an asset acquisition.
The performance rights associated with the development milestones have been recognized as an equity settled share-
based payment at a fair value of $23,160,000 which has been included in the fair value of intellectual property. Each
milestone associated with the rights has a fixed dollar amount which can be settled either in cash or shares. The fair
value of the performance rights was determined based on management’s assessment of the likelihood of each milestone
being reached against the fixed dollar amount for that milestone. The likelihood of the milestones being attained are
considered non-vesting conditions as there are no further services or obligations of the counterparty, thus being
reflected in the fair value.
assumed at the acquisition date.
F-42
Fair value | |||
Consideration | US$'000 | 1. | |
Cash paid | |||
Performance rights issued | |||
Equity issued | |||
Acquisition related costs | |||
Deferred payment | |||
Total consideration | |||
Recognized amounts of identifiable assets acquired and liabilities assumed | |||
Property, plant and equipment | |||
Right-of-use assets | |||
Intellectual property | |||
Licenses | |||
Lease liabilities | ( | ||
Total identifiable assets and liabilities |
Acquisition-related intangible assets of $25,866,000 relate to the valuation of the acquired ImaginAb therapeutic
candidate intellectual property and $41,145,000 associated with a perpetual license for the targeting technology
platform. The useful economic life of the intellectual property has not been assessed at the acquisition date, as the
intellectual property is not available for commercial use until regulatory approval has been obtained.
23.3 Acquisition of FAP targeting candidates
On March 12, 2025 Telix entered into an asset purchase and exclusive worldwide in-license agreements for a suite of
clinically validated FAP-targeting therapeutic and precision medicine (diagnostic) radiopharmaceutical candidates.
At closing Telix entered into an exclusive worldwide license agreement with a German company controlled by Professor
Frank Roesch, SCV GmbH, and a concurrently-signed asset purchase agreement with German company Medianezia
GmbH, which collectively hold the intellectual property rights to the FAP assets.
Telix paid €5,300,000 (US$5,774,000 ) in cash (in addition to €700,000 (US$747,000 ) paid upfront at agreement
signing); and will pay a further €4,000,000 (US$4,354,000 ) in cash in the 12 months following entry into the agreements,
subject to potential indemnity set-off. Telix will pay up to a further €132,000,000 (US$154,651,200 ) contingent upon
achievement of certain clinical development and regulatory milestones related to both the diagnostic and therapeutic
candidates under both agreements. An additional €20,000,000 (US$23,432,000 ) will be payable under the license
agreement on achievement of certain commercial milestones related to the diagnostic product; as well as royalties on net
sales in the low to mid-single digits on the diagnostic product and an earlier formulation of the therapeutic product, if
used.
The Group has assessed that the acquisition does not meet the definition of a business in IFRS 3/AASB 3 Business
Combinations as the Group concluded that the acquisition does not include a self-sustaining substantive process
capable of generating outputs. On this basis, the Group has concluded that the components acquired will be treated as
an asset acquisition.
Contingent consideration in connection with the purchase of individual assets outside of business combinations is
recognized as a financial liability only when the non-contingent obligation arises (i.e. when milestone is met). The
determination of whether the payment should be capitalized or expensed is usually based on the reason for the
contingent payment.
For the acquisition of the FAP targeting candidates, contingent payments based on regulatory approvals received (i.e.
development milestones) will be capitalized as the payments are incidental to the acquisition and making the asset
available for its intended use. Contingent payments associated with sales related milestones will be expensed.
F-43
Fair value | |||
Consideration | US$'000 | 1. | |
Cash paid | |||
Acquisition related costs | |||
Deferred payment | |||
Total consideration | |||
Recognized amounts of identifiable assets acquired and liabilities assumed | |||
Intellectual property | |||
License agreement | |||
Total identifiable assets and liabilities |
23.4 Acquisition of ARTMS, Inc.
On 11 April 2024 Telix completed the acquisition of radioisotope production technology firm ARTMS, Inc. (ARTMS). During
the period the Group finalised the purchase price allocation associated with the ARTMS acquisition. The adjustments to
the acquired assets and liabilities and resultant goodwill were as follows:
Fair value | ||||
Identifiable assets and liabilities | Increase or decrease | US$'000 | ||
Intellectual property | Increase | |||
Deferred tax liabilities | Increase | ( | ||
Goodwill | Decrease |
24. Trade and other payables
2025 | 2024 | |||
US$'000 | US$'000 | |||
(Recast) | ||||
Trade creditors | ||||
Accruals | ||||
Deferred consideration | ||||
Customer rebates payable | ||||
Other creditors | ||||
Accrued royalties | ||||
Payroll liabilities | ||||
Government rebates payable | ||||
Total trade and other payables |
F-44
25. Borrowings
2025 | 2024 | |||||||
Current | Non-current | Current | Non-current | 1. | ||||
US$'000 | US$'000 | US$'000 | US$'000 | 1. | ||||
(Recast) | (Recast) | 1. | ||||||
Secured | ||||||||
Bank loans | ||||||||
Working capital facility | ( | ( | ||||||
Total secured borrowings | ||||||||
Unsecured | ||||||||
Convertible bonds | ||||||||
Total unsecured borrowings | ||||||||
Total borrowings | ||||||||
Loan balance | Due < 1 year | Due > 1 year | Facility limit | Maturity date | 1. | |||||
Lenders | US$'000 | US$'000 | US$'00 0 | US$'000 | 1. | |||||
The Hongkong and Shanghai Banking Corporation Limited As The Trustee For Convertible Bond Holders | 30-Jul-29 | |||||||||
IMBC Group | 31-Mar-33 | |||||||||
BNP Paribas | 29-Feb-32 | |||||||||
HSBC Bank Australia Ltd | ( | ( | utilization | |||||||
Total |
December 31, 2024
Loan balance | Due < 1 year | Due > 1 year | Facility limit | Maturity date | 1. | |||||
US$'000 | US$'000 | US$'000 | US$'000 | 1. | ||||||
Lenders | (Recast) | (Recast) | (Recast) | (Recast) | 1. | |||||
The Hongkong and Shanghai Banking Corporation Limited As The Trustee For Convertible Bond Holders | 30-Jul-29 | |||||||||
IMBC Group | 31-Mar-33 | |||||||||
BNP Paribas | 29-Feb-32 | |||||||||
HSBC Bank Australia Ltd | ( | ( | utilization | |||||||
Total |
F-45
The bank loans outstanding at December 31, 2025 are in relation to the build-out of the Brussels South
radiopharmaceutical production facility. Telix Pharmaceuticals (Belgium) SPRL (a wholly owned subsidiary of Telix)
entered into two loan agreements, one with BNP Paribas and IMBC Group totaling €10,100,000 on a 10-year term, and a
second loan with BNP Paribas totaling €2,000,000 on a two-year extendable term. All loans have a two-year repayment
holiday period, with repayments due to commence from March 2024. The loans are secured by a fixed charge over the
facility.
The loan agreements entitle BNP Paribas and IMBC Group to suspend or terminate all or part of the undrawn portion of
the loan facilities with immediate effect and without prior notice. At December 31, 2025, the undrawn portion under the
agreements was €1,989,000 ($2,330,000 ). As at the reporting date Telix has not received any notice to this effect.
The loan agreements require Telix Pharmaceuticals (Belgium) SRL to comply with various covenants relating to the
conduct of the business, including non-payment of required repayments, specified cross-defaults (in the event of the
use of trade bills) and ensuring cumulative losses of Telix Pharmaceuticals (Belgium) SPRL do not exceed 25 % of its
capital and reserves. Upon the occurrence of an event of default and in the event of a change of control, BNP Paribas
and IMBC Group may accelerate payments due under the loan agreements or terminate the loan agreements. There were
no events of default or changes of control during the year.
On December 17, 2024, the Group entered into an agreement with HSBC Bank Australia Limited ("HSBC") to obtain a
working capital facility of up to $33,415,000 (A$50,000,000 ). To date, the Group has not utilized this facility and has
incurred establishment fee costs of $99,000 (A$150,000 ) associated with the facility.
The working capital facility is secured by a cash security deposit on an interest-bearing term deposit of $33,415,000
(A$50,000,000 ) held by HSBC with a maturity date equivalent to the term of the facility. There are no financial covenants
associated with the facility. Refer to note 17 for further details.
On July 30, 2024 the Group completed the issue of $426,140,000 (A$650,000,000 ) in convertible bonds maturing in
2029. The bonds are convertible into fully paid ordinary shares in Telix Pharmaceuticals Limited. The initial conversion
price of the convertible bonds is A$24.78 per share, subject to anti-dilution adjustments set out in the final terms and
conditions of the convertible bonds. The net proceeds were $416,324,000 , after transaction costs.
The convertible bonds will bear interest at a rate of 2.375 per cent per annum. Interest will be payable quarterly in
arrears on October 30, January 30, April 30 and July 30 in each year, beginning on October 30, 2024. The convertible
bonds will mature on or about July 30, 2029, unless redeemed, repurchased, or converted in accordance with their
terms. The holders have the option to redeem all or some of the convertible bonds on July 30, 2027 at their principal
amount together with accrued but unpaid interest accrued to that date. The convertible bonds are listed on the SGX.
2025 | 2024 | 1. | ||
US$'000 | US$'000 | 1. | ||
(Recast) | ||||
Opening balance | ||||
Face value of convertible bonds issued | ||||
Transaction costs | ( | |||
Other equity securities - value of conversion rights | ( | |||
Unwind of discount | ||||
Interest expense | ||||
Interest paid | ( | ( | ||
Exchange differences | ( | |||
Closing balance | ||||
Current | ||||
Non-current | ||||
Total convertible bond liability |
F-46
The initial fair value of the liability portion of the bond was determined using a market interest rate for an equivalent non-
convertible bond at the issue date. This fair value has been reduced by directly attributable transaction costs associated
with the issue of the convertible bonds. The liability is subsequently recognized on an amortized cost basis until
extinguished on conversion or maturity of the bonds. The remainder of the proceeds is allocated to the conversion option
and recognized as part of the share capital reserve, net of income tax and a proportion of transaction costs, and is not
subsequently remeasured. Refer to note 30.2.2 for further details.
Opening balance | Net cash inflow/ (outflow) | Other non- cash movements | Closing balance | 1. | ||||
US$'000 | US$'000 | US$'000 | US$'000 | 1. | ||||
For the year ended December 31, 2025 | ||||||||
Bank loans | ( | |||||||
Working capital facility | ( | ( | ( | |||||
Convertible bonds | ( | |||||||
Lease liabilities | ( | |||||||
( | ||||||||
(Recast) | (Recast) | (Recast) | (Recast) | |||||
For the year ended December 31, 2024 | ||||||||
Bank loans | ( | |||||||
Working capital facility | ( | ( | ||||||
Convertible bonds | ( | |||||||
Lease liabilities | ( | |||||||
( |
Other non-cash movements include recognition of the conversion option as part of the share capital reserve, new leases
entered into during the year, leases acquired via acquisitions of a business, disposal of leases and exchange differences.
For bank loans, the fair values are not materially different to their carrying amounts, since the interest payable on those
borrowings is either close to current market rates or the borrowings are of a short-term nature.
current borrowing rate. They are classified as level 3 fair values in the fair value hierarchy (refer to note 33.6) due to the
use of unobservable inputs, including own credit risk.
2025 | 2024 | 1. | ||||||
Carrying amount | Fair value | Carrying amount | Fair value | 1. | ||||
US$'000 | US$'000 | US$'000 | US$'000 | 1. | ||||
(Recast) | (Recast) | 1. | ||||||
Bank loans | ||||||||
Convertible bonds | ||||||||
Capital risk management: Capital is defined as the combination of shareholders’ equity, reserves and net debt. The key
objective of the Group when managing its capital is to safeguard its ability to continue as a going concern, so that the
Group can continue to provide benefits for stakeholders and maintain an optimal capital and funding structure. The aim
of the Group’s capital management framework is to maintain, monitor and secure access to future funding arrangements
F-47
26. Lease liabilities
2025 | 2024 | |||
US$'000 | US$'000 | |||
(Recast) | ||||
Balance at January 1, | ||||
Additions | ||||
Acquisition of businesses | ||||
Interest expense | ||||
Lease payments (principal and interest) | ( | ( | ||
Exchange differences | ( | |||
Balance at December 31, |
Lease liabilities | 2025 | 2024 | 1. | |
US$'000 | US$'000 | 1. | ||
(Recast) | 1. | |||
Current | ||||
Non-current | ||||
Total lease liabilities |
Interest expense relating to leases | 2025 | 2024 | 2023 | |||
US$'000 | US$'000 | US$'000 | ||||
(Recast) | (Recast) | 1. | ||||
Properties | ||||||
Motor vehicles | ||||||
Total lease interest |
The total cash outflow for leases in 2025 comprises $5,237,000 (2024: $1,317,000 , 2023: $1,266,000 ) principal and
$3,637,000 (2024: $491,000 , 2023: $423,000 ) interest payments.
F-48
27. Provisions
Government grant liability | Decommissioning liability | Total | 1. | |||
US$'000 | US$'000 | US$'000 | ||||
Balance at January 1, 2025 | ||||||
Remeasurement of provisions | ( | ( | ||||
Unwind of discount | ||||||
Charged to profit or loss | ||||||
Exchange differences | ||||||
Amounts adjusted to property, plant and equipment | ( | ( | ||||
Provision utilized | ( | ( | ( | |||
Balance at December 31, 2025 | ||||||
Current | ||||||
Non-current | ||||||
Total provisions | ||||||
(Recast) | (Recast) | (Recast) | ||||
Balance at January 1, 2024 | ||||||
Remeasurement of provisions | ||||||
Unwind of discount | ||||||
Charged to profit or loss | ||||||
Exchange differences | ( | ( | ( | |||
Amounts adjusted to property, plant and equipment | ||||||
Provision utilized | ( | ( | ( | |||
Balance at December 31, 2024 | ||||||
Current | ||||||
Non-current | ||||||
Total provisions |
Telix Innovations has received grants from the Walloon regional government in Belgium. These grants meet the definition
of a financial liability as defined in IFRS 9 Financial Instruments and were designated to be measured at fair value through
profit and loss.
The grants are repayable to the Walloon government based on a split between fixed and variable repayments. The fixed
proportion is based on contractual cash flows agreed with the Walloon government. The variable cash flows are based
on a fixed percentage of future sales and are capped at an agreed upon level.
The Group has estimated that the full variable repayments will be made up to the pre-agreed capped amount. The key
inputs into this calculation are the risk adjusted discount rate of 3.9 % (2024: 3.3 %), the expected sales volumes and the
net sales price per unit. The expected sales volumes and net sales price per unit assumptions are consistent with those
utilized by the Group in the calculation of the contingent consideration liability and intellectual property valuation.
Telix owns and operates a radiopharmaceutical production facility in Belgium. The site has cyclotrons installed in
concrete shielded vaults which also contain some nuclear contamination associated with past manufacturing activities.
Telix has an obligation to remove the cyclotrons and restore the site.
In 2024, new cyclotrons were installed in the facility, which will be decommissioned at the end of the operating life of the
facility. A provision for dismantling and removal of $3,656,000 has been recognized with respect to these cyclotrons, in
addition to existing remediation costs to remove nuclear contamination in the vaults.
F-49
The total decommissioning costs expected to be incurred in 2041 of €12,451,000 (2024: €12,451,000 ) have been
discounted using the Belgium risk-free rate of 3.9 % (2024: 3.3 %) and translated to US$ at the exchange rate at
December 31, 2025.
The provision represents the best estimate of the expenditures required to settle the present obligation at December 31,
2025. While the Group has made its best estimate in establishing its decommissioning liability, because of potential
changes in technology as well as safety and environmental requirements, plus the actual timescale to complete
decommissioning, the ultimate provision requirements could vary from the Group’s current estimates. Any subsequent
changes in estimate which alter the level of the provision required are also reflected in adjustments to the intangible
license asset. Each year, the provision is increased to reflect the unwind of discount and to accrue an estimate for the
effects of inflation, with the charges being presented in the consolidated statement of comprehensive income or loss.
Actual payments for commencement of decommissioning activity are disclosed as provision utilized in the above table.
28. Contingent consideration
US$'000 | |
Balance at January 1, 2025 | |
Remeasurement of contingent consideration | ( |
Unwind of discount | |
Charged to profit or loss | ( |
Exchange differences | |
Acquisition of businesses | |
Amounts adjusted to intangible assets | ( |
Payments for contingent consideration (Operating) | ( |
Payments for contingent consideration (Investing) | ( |
Balance at December 31, 2025 | |
Current | |
Non-current | |
Total contingent consideration | |
(Recast) | |
Balance at January 1, 2024 | |
Remeasurement of contingent consideration | |
Unwind of discount | |
Charged to profit or loss | |
Exchange differences | ( |
Acquisition of businesses | |
Amounts adjusted to intangible assets | |
Payments for contingent consideration (Operating) | ( |
Payments for contingent consideration (Investing) | ( |
Balance at December 31, 2024 | |
Current | |
Non-current | |
Total contingent consideration |
28.1. Telix Innovations (formerly ANMI)
The Group acquired ANMI on December 24, 2018. The Group is liable for future variable payments which are calculated
based on the percentage of net sales for five years following the achievement of marketing authorization of the product.
The percentage of net sales varies depending on the net sales achieved in the U.S. and the rest of the world. The Group
also held and exercised an option to buy-out the remaining future variable payments during the year, as the specified
sales thresholds were met.
F-50
As at consolidated statement of financial position date, the Group remeasured the contingent consideration by
$1,973,000 as a result of actual sales volumes and fully settled the outstanding balance of $51,657,000 .
28.2. Telix Switzerland (formerly TheraPharm)
Telix acquired TheraPharm on December 14, 2020. Part of the consideration for the acquisition was in the form of future
payments contingent on certain milestones. These are:
•€5,000,000 cash payment upon successful completion of a Phase 3 pivotal registration trial
•€5,000,000 cash payment upon achievement of marketing authorization in Europe or the U.S., whichever approval
comes first, and
•5 % of net sales for the first three years following marketing authorization in Europe or the U.S., whichever approval
comes first.
The valuation of the contingent consideration has been performed utilizing a discounted cash flow model that uses
certain unobservable assumptions. These key assumptions include risk adjusted post-tax discount rate of 13.2 % (2024:
approval for marketing authorization probability success factor.
sensitivities from reasonably possible changes where applicable:
Contingent consideration valuation
1. | Unobservable input | Methodology | December 31, 2025 | ||
Risk adjusted post- tax discount rate | The post-tax discount rate used in the valuation has been determined based on required rates of returns of listed companies in the biotechnology industry (having regards to their stage of development, size and risk adjustments). | A discount rate would decrease / increase the contingent consideration by $ | |||
Expected sales volumes | This is determined through assumptions on target market population, penetration and growth rates in the United States and Europe. | A volumes would increase / decrease the contingent consideration by $ | |||
Net sales price per unit | The net sales price per unit is estimated based on comparable products currently in the market. | A price per unit would increase / decrease the contingent consideration by $ | |||
Approval for marketing authorization probability success factor | This assumption is based on management’s estimate for achieving regulatory approval and is determined through benchmarking of historic approval rates. | An increase / decrease in the probability of success factor by decrease the contingent consideration by $ |
28.3. IsoTherapeutics
The Group acquired IsoTherapeutics on April 9, 2024. The Group is liable for $5,000,000 which is payable in cash for
performance-related milestone payments that are subject to meeting milestone conditions within twelve months of
closing. During the year ended December 31, 2025, the milestone conditions were satisfied and the associated liability
was settled.
28.4. ARTMS
Telix acquired ARTMS on April 11, 2024. Part of the consideration for the acquisition included US$24.5 million in
contingent future earn-out payments which is payable in cash following achievement of certain clinical or commercial
milestones. All earn-outs which have not otherwise expired will terminate on the 10 year anniversary of completion.
In addition to the above, the contingent consideration includes future royalty payments for a low single to low double-
digit percentage of net sales of ARTMS products or Telix products.
The contingent consideration liability has been valued using a discounted cash flow model that utilizes certain
unobservable level 3 inputs. These key assumptions include risk adjusted post-tax discount rate of 14.3 % (2024: 15.0 %),
FDA approval dates, expected sales volume over the forecast period and net sales price per unit and a probability
success factor in relation to ARTMS achieving its clinical or commercial milestones.
F-51
sensitivities from reasonably possible changes where applicable:
Contingent consideration valuation
1. | Unobservable input | Methodology | December 31, 2025 | ||
Risk adjusted post- tax discount rate | The post-tax discount rate used in the valuation has been determined based on required rates of returns of listed companies in the biotechnology industry (having regards to their stage of development, size and risk adjustments). | A discount rate would decrease / increase the contingent consideration by $ | |||
Expected sales volumes - ARTMS and Telix products | This is determined through assumptions on target market population, penetration and growth rates in the United States and Europe. | A volumes would increase / decrease the contingent consideration by $ | |||
Net sales price per unit | The net sales price per unit is estimated based on comparable products currently in the market. | A price per unit would increase / decrease the contingent consideration by $ across the different royalties. | |||
Milestone achievement probability of success factor | This assumption is based on management’s estimate for achieving the clinical or commercial milestones. | An increase / decrease in the probability of success factor by decrease the contingent consideration by $ |
29. Employee benefit obligations
2025 | 2024 | |||
US$'000 | US$'000 | |||
(Recast) | ||||
Bonuses | ||||
Annual leave | ||||
Long service leave | ||||
Balance at December 31, | ||||
Current | ||||
Non-current | ||||
Total employee benefit obligations |
F-52
30. Equity
30.1. Share capital
2025 | 2024 | 2023 | 2025 | 2024 | 2023 | |||||||
Number ‘000 | Number ‘000 | Number ‘000 | US$'000 | US$'000 | US$'000 | 1. | ||||||
(Recast) | (Recast) | |||||||||||
Balance at January 1, | ||||||||||||
Shares issued through the exercise of share options and warrants1 | ||||||||||||
Shares issued for Dedicaid2 | ||||||||||||
Shares issued for Lightpoint3 | ||||||||||||
Shares issued for IsoTherapeutics4 | ||||||||||||
Shares issued for ARTMS5 | ||||||||||||
Shares issued for QSAM6 | ||||||||||||
Shares issued for Imaginab7 | ||||||||||||
Transaction costs arising on new share issues | ||||||||||||
Balance at December 31, |
1.Options exercised during the year through the employee Equity Incentive Plan resulted in 1,693,000 (2024: 525,000 ,
2023: 3,879,000 ) shares being issued of total value of $29,722,000 (2024: $5,357,000 , 2023: $27,857,000 ).
2.On April 27, 2023, the Group completed the acquisition of Dedicaid GmbH. The consideration for the acquisition
comprised 207,000 in Telix shares at a 10-day volume weighted average price of shares on the execution date of
A$8.73 per share. During the year, the Group issued 37,575 fully paid ordinary shares upon satisfaction of certain
milestones.
3.On November 1, 2023, the Group completed the acquisition of Lightpoint through the issue of 3,298,000 fully paid
ordinary Telix shares at A$9.37 per share. During the year, the Group issued 269,075 fully paid ordinary shares in
satisfaction of Lightpoint milestone rights.
4.On April 9, 2024, the Group completed the acquisition of IsoTherapeutics. The consideration included the issue of
5.On April 11, 2024, the Group completed the acquisition of ARTMS. The consideration included the issue of 5,674,365
fully paid ordinary Telix shares at A$12.62 per share.
6.On May 3, 2024, the Group completed the acquisition of QSAM. The purchase price included the issue of 3,671,120
fully paid ordinary Telix shares at $14.80 per share and a further 409,026 fully paid ordinary Telix shares at $18.05 per
share.
7.On January 30, 2025, the Group completed the acquisition of ImaginAb. The consideration included the issue of
The weighted average ordinary shares for the period January 1, 2025 to December 31, 2025 is 337,879,901 (2024:
law.
Rights applying to securities:
1.Ordinary shares: Ordinary shares entitle the holder to participate in dividends, and to share in the proceeds of winding
up the Company in proportion to the number of and amounts paid on the shares held.
2.Options and rights: Holders of Options and rights have no voting rights. Information relating to the Company’s
Employee Incentive Plan ("EIP"), including details of Options issued, exercised and lapsed during the financial year, is
set out in note 31.
F-53
30.2. Share capital reserve
2025 | 2024 | 2023 | 2025 | 2024 | 2023 | |||||||
Number ’000 | Number ’000 | Number ‘000 | US$'000 | US$'000 | US$'000 | 1. | ||||||
(Recast) | (Recast) | |||||||||||
Balance at January 1 | ( | ( | ||||||||||
Treasury shares acquired | ( | ( | ( | |||||||||
Issue of convertible bonds | ||||||||||||
Transaction costs arising on convertible bonds issue | ( | |||||||||||
Shares allocated to employees | ( | ( | ( | |||||||||
Balance at December 31 | ( | ( |
30.2.1. Treasury shares
Ordinary shares in the Company were purchased by the Telix Pharmaceuticals Employee Share Trust for the purpose of
issuing shares under the Equity Incentive Plan. These shares are allocated to employees and are not held within the
Employee Share Trust (see note 31 for further information).
30.2.2. Conversion right of convertible bonds
The amount shown for the issue of convertible bonds is the initial value of the conversion rights relating to the
convertible bonds.
30.3. Other reserves
Foreign currency translation reserve | Share-based payments reserve | Financial assets at FVOCI reserve | Total | 1. | ||||
US$'000 | US$'000 | US$'000 | US$'000 | |||||
Balance as at January 1, 2025 | ( | ( | ||||||
Other comprehensive (loss)/income | ( | ( | ( | |||||
Total comprehensive (loss)/income | ( | ( | ( | |||||
Share-based payments to employees | ||||||||
Share-based payments associated with acquisitions | ||||||||
Transfer on satisfaction of acquisition performance rights | ( | ( | ||||||
Transfer on exercise of options | ( | ( | ||||||
Balance as at December 31, 2025 | ( | ( |
F-54
Foreign currency translation reserve | Share-based payments reserve | Financial assets at FVOCI reserve | Total | 1. | ||||
US$'000 | US$'000 | US$'000 | US$'000 | |||||
(Recast) | (Recast) | (Recast) | (Recast) | |||||
Balance as at January 1, 2024 | ( | ( | ||||||
Other comprehensive | ( | |||||||
Total comprehensive income/(loss) | ( | |||||||
Share-based payments to employees | ||||||||
Share-based payments associated with acquisitions | ||||||||
Transfer on exercise of options | ( | ( | ||||||
Balance as at December 31, 2024 | ( | ( |
Foreign currency translation reserve | Share-based payments reserve | Financial assets at FVOCI reserve | Total | 1. | ||||
US$'000 | US$'000 | US$'000 | US$'000 | |||||
(Recast) | (Recast) | (Recast) | (Recast) | |||||
Balance as at January 1, 2023 | ( | |||||||
Other comprehensive (loss)/income | ( | ( | ( | |||||
Total comprehensive (loss)/income | ( | ( | ( | |||||
Share-based payments to employees | ||||||||
Share-based payments associated with acquisitions | ||||||||
Transfer on exercise of options | ( | ( | ||||||
Balance as at December 31, 2023 | ( | ( |
30.4. Share-based payments reserve
2025 | 2024 | 2023 | ||||
Number ’000 | Number ’000 | Number ‘000 | ||||
Balance at January 1 | ||||||
EIP options issued | ||||||
Options exercised | ( | ( | ( | |||
Options lapsed | ( | ( | ( | |||
Performance Rights issued1 | ||||||
Performance Rights exercised | ( | |||||
Performance Rights lapsed | ( | |||||
Balance at December 31 |
1.Relates to the acquisition of ImaginAb in 2025, QSAM in 2024 and Lightpoint in 2023.
1 WAEP - weighted average exercise price
F-55
30.5. Financial assets at FVOCI reserve
The group has elected to recognize changes in the fair value of certain investments in equity securities in Other
comprehensive income (OCI), as explained in note 17. These changes are accumulated within the FVOCI reserve within
equity.
2025 | 2024 | 2023 | ||||
US$'000 | US$'000 | US$'000 | 1. | |||
(Recast) | (Recast) | 1. | ||||
Balance at January 1 | ( | ( | ||||
Revaluation - gross | ( | ( | ( | |||
Deferred tax | ||||||
Balance at December 31 | ( | ( | ( |
31. Share-based payments
Equity Incentive Plan and Options
The Equity Incentive Plan ("EIP") was established to allow the Board of Telix to make offers to Eligible Employees to
acquire securities in the Company and to otherwise incentivize employees. ‘Eligible Employees’ includes full time, part
time or casual employees of a Group Company, a Non-Executive Director of a Group Company, a Contractor, or any
other person who is declared by the Board to be eligible.
The Board may, from time to time and in its absolute discretion, invite Eligible Employees to participate in a grant of
Incentive Securities, which may comprise Rights (including Performance Share Appreciation Rights), Options, and/or
Restricted Shares. Vesting of Incentive Securities under the EIP is subject to any vesting or performance conditions
determined by the Board. Incentive Securities are normally granted under the EIP for no consideration and carry no
dividend or voting rights. When exercised, each Incentive Security is convertible into one ASX or ADS-backed Share.
Non-Executive Directors are able to participate in the Equity Incentive Plan, under which equity may be issued subject to
Shareholder approval. Options were issued to Non-Executive Directors not as an ‘incentive’ under the EIP but as a means
of cost-effective consideration for agreeing to join the Board. The details of Incentive Securities on issue to individual
Directors can be found in the Remuneration report for the year ended December 31, 2025. For the purposes of this table
and to illustrate the total number of Incentive Securities on issue under the rules of the EIP, all Incentive Securities issued
to Non-Executive Directors, Executive Directors, employees and contractors are included.
Incentive Securities contain a cashless exercise clause that allows employees to exercise the securities for a cash
exercise price of $nil in exchange for forfeiting a portion of their vested securities.
ASX securities
2025 | 2025 | 2024 | 2024 | |||||
Number | WAEP11 | Number | WAEP1 | 1. | ||||
‘000 | AU$ | ‘000 | AU$ | 1. | ||||
Balance at January 1, | ||||||||
Granted during the year | ||||||||
Exercised during the year | ( | ( | ||||||
Lapsed/forfeited during the year | ( | ( | ||||||
Balance at December 31, | ||||||||
Vested and exercisable at December 31, |
F-56
ADS-backed securities
2025 | 2025 | 2024 | 2024 | ||||
Number | WAEP1 | Number | WAEP1 | ||||
‘000 | US$ | ‘000 | US$ | ||||
Balance at January 1, | |||||||
Granted during the year | |||||||
Exercised during the year | ( | ||||||
Lapsed/forfeited during the year | ( | ||||||
Balance at December 31, | |||||||
Vested and exercisable at December 31, |
2025 | 2024 | 2023 | ||||
US$ | US$ | US$ | 1. | |||
(Recast) | (Recast) | 1. | ||||
Options issued under EIP | ||||||
Total |
F-57
Equity Incentive Plan and Options
Grant date | Vesting date | Expiry date | Exercise price | Options on issue at January 1, 2025 | Issued during the year | Vested during the year | Exercised during the year | Lapsed during the year | Options on issue at December 31, 2025 |
A$ | ’000 | ’000 | ’000 | ’000 | ’000 | ’000 | |||
01-27-2021 | 10-28-2022 | 01-26-2026 | ( | ||||||
07-27-2021 | 10-28-2022 | 07-27-2026 | ( | ||||||
07-27-2021 | 07-27-2025 | 07-27-2026 | |||||||
04-05-2022 | 12-31-2024 | 04-04-2027 | ( | ( | |||||
04-05-2022 | 12-31-2024 | 04-04-2027 | ( | ||||||
10-24-2022 | 12-31-2024 | 10-24-2027 | ( | ||||||
05-02-2023 | 12-31-2025 | 03-27-2028 | ( | ||||||
21-Mar-24. 22-May-24 | 12-31-2025 | 05-16-2028 | ( | ||||||
07-06-2023 | 31-Mar-25 or 31-Dec-25 | 15-Jun-25, 15-Jun-28 | ( | ||||||
10-18-2023 | 06-30-2026 | 09-20-2028 | ( | ||||||
10-31-2023 | 12-31-2026 | 10-31-2028 | ( | ||||||
10-31-2023 | 12-31-2027 | 10-31-2029 | ( | ||||||
11-30-2023 | 06-30-2026 | 11-14-2028 | ( | ||||||
03-08-2024 | 12-31-2026 | 03-31-2029 | |||||||
03-08-2024 | 12-31-2027 | 03-31-2030 | |||||||
21-Mar-24, 22-May-24 | 03-31-2027 | 03-31-2029 | ( | ||||||
04-26-2024 | 03-31-2027 | 03-31-2029 | |||||||
08-26-2024 | 04-01-2025 | 03-31-2027 | ( | ||||||
08-26-2024 | 03-31-2027 | 04-04-2027 | |||||||
09-19-2024 | 03-31-2028 | 03-31-2029 | ( | ||||||
09-19-2024 | 03-31-2028 | 03-31-2030 | |||||||
10-17-2024 | 11-01-2027 | 11-01-2029 | ( | ||||||
10-17-2024 | 11-01-2028 | 11-01-2030 | ( | ||||||
01-01-2025 | 03-31-2028 | 03-31-2030 | ( | ||||||
01-01-2025 | 02-28-2026 | 03-31-2028 | |||||||
10-24-2025 | 03-31-2028 | 03-31-2028 | ( | ||||||
03-17-2025 | 09-01-2025 | 09-30-2025 | ( | ||||||
03-17-2025 | 03-01-2026 | 03-15-2026 | ( | ||||||
( | ( |
F-58
Details of the number of options convertible to ADS-backed shares issued under the EIP outstanding at the end of the
year:
Grant date | Vesting date | Expiry date | Exercise price | Options on issue at January 1, 2025 | Issued during the year | Vested during the year | Exercised during the year | Lapsed during the year | Options on issue at December 31, 2025 |
US$ | ’000 | ’000 | ’000 | ’000 | ’000 | ’000 | |||
01-01-2025 | 03-31-2028 | 03-31-2030 | ( | ||||||
01-01-2025 | 02-27-2026 | 03-13-2026 | |||||||
10-24-2025 | 03-31-2028 | 03-31-2028 | |||||||
03-17-2025 | 09-01-2025 | 09-30-2025 | ( | ( | |||||
03-17-2025 | 03-01-2026 | 03-15-2026 | ( | ||||||
( | ( |
independently determined using the Black Scholes Model. The model inputs for options granted during the year ended
December 31, 2025 are included below:
ASX securities
January 2025 | January 2025 | May 2025 | October 2025 | March 2025 | March 2025 | ||||||
Fair value | A$ | A$ | A$ | A$ | A$ | A$ | |||||
Consideration | |||||||||||
Exercise price | A$ | A$ | A$ | A$ | A$ | A$ | |||||
Grant date | 01-01-2025 | 01-01-2025 | 05-21-2025 | 10-24-2025 | 03-17-202 5 | 03-17-202 5 | |||||
Expiry date | 03-31-2030 | 03-31-202 8 | 03-31-2028 | 03-31-2028 | 09-30-202 5 | 03-15-202 6 | |||||
Term | |||||||||||
Share price at grant date | A$ | A$ | A$ | A$ | A$ | A$ | |||||
Volatility | |||||||||||
Dividend yield | |||||||||||
Risk-free rate |
ADS-backed securities
January 2025 | January 2025 | October 2025 | March 2025 | March 2025 | |||||
Fair value | US$ | US$ | US$ | US$ | US$ | ||||
Consideration | |||||||||
Exercise price | US$ | US$ | US$ | US$ | US$ | ||||
Grant date | 01-01-2025 | 01-01-2025 | 10-24-202 5 | 03-17-2025 | 03-17-2025 | ||||
Expiry date | 03-31-2030 | 03-13-2026 | 03-31-202 8 | 09-30-2025 | 03-15-2026 | ||||
Term | |||||||||
Share price at grant date | A$ | A$ | A$ | A$ | A$ | ||||
Volatility | |||||||||
Dividend yield | |||||||||
Risk-free rate |
F-59
32. Cash flow information
32.1. Reconciliation of (loss)/profit after income tax to net cash (used in)/from operating activities
2025 | 2024 | 2023 | ||||
US$'000 | US$'000 | US$'000 | ||||
(Recast) | (Recast) | |||||
(Loss)/profit before income tax | ( | |||||
Adjustments for | ||||||
Depreciation and amortization | ||||||
Impairments/(impairment reversals) of intangible assets | ( | |||||
Fair value remeasurement of contingent consideration | ( | |||||
Fair value remeasurement of provisions | ( | ( | ||||
Unwind of discount | ||||||
Share-based payments | ||||||
Foreign exchange (losses)/gains | ( | ( | ||||
Interest paid | ( | ( | ( | |||
Income taxes paid | ( | ( | ( | |||
Change in assets and liabilities | ||||||
Increase in trade and other receivables | ( | ( | ( | |||
Increase in inventory | ( | ( | ( | |||
Decrease/(increase) in other current assets | ( | ( | ||||
Decrease/(increase) in other non-current assets | ( | |||||
Increase in trade creditors | ||||||
Deduct trade and other payables capitalized to intangible assets | ( | |||||
Contingent consideration payments classified as operating | ( | ( | ( | |||
Increase in employee benefit obligations | ||||||
(Decrease)/Increase in provisions | ( | ( | ||||
Decrease in contract liabilities | ( | ( | ( | |||
Net cash (used in)/from operating activities | ( |
33. Financial risk management
The Group’s activities expose it to a variety of financial risks: market risk, credit risk and liquidity risk. The overall risk
management program focuses on the unpredictability of markets and seeks to minimize potential adverse effects on the
financial performance of the Group. The Group uses different methods to measure different types of risk to which it is
exposed.
33.1. Interest rate risk
The Group’s borrowings that have been drawn down at December 31, 2025 have fixed interest rates, and therefore the
Group is not exposed to any significant interest rate risk.
33.2. Price risk
The Group's exposure to equity securities price risk arises from investments held by the Group and classified in the
consolidated statement of financial position at fair value through other comprehensive income ("FVOCI") (note 17).
The amounts recognized in other comprehensive income in relation to investments held by the Group are disclosed in
note 30.5.
F-60
33.3. Foreign currency risk
The Group operates internationally and is exposed to foreign exchange risk, primarily the Australian dollar and Euro.
Foreign exchange risk arises from commercial activities outside the U.S. and research and development activities in
Europe.
The Group’s treasury risk management policy is to settle all US dollar denominated expenditure with US dollar
denominated receipts from sales of Illuccix in the U.S. The Group also manages currency risk by making decisions as to
the levels of cash to hold in each currency by assessing its future activities which will likely be incurred in those
currencies. Any remaining foreign currency exposure has therefore not been hedged.
We have both foreign currency receivables and payables, predominantly denominated in Australian dollars and Euros. We
had a surplus of foreign currency receivables and financial assets over payables of $67.4 million as of December 31,
2025 (December 31, 2024: $49.6 million).
The Group’s exposure to the risk of changes in foreign exchange rates also relates to the Group’s net investments in
foreign subsidiaries, which predominantly include denominations in Euro and Australian dollar, however given the level of
current investments in non US dollar denominated subsidiaries, the impact is limited.
As at December 31, 2025, the Group held 52.8 % (2024: 32.0 %) of its cash in Australian dollars, 39.7 % (2024: 64.9 %) in
US dollars, 5.5 % (2024: 2.8 %) in Euros (EUR), 0.1 % (2024: nil ) in Japanese Yen (JPY), 0.3 % (2024: 0.1 %) in British pounds,
Exposure
As at December 31, 2025 | |||||||||||||
USD | AUD | EUR | CHF | JPY | GBP | CAD | |||||||
US$'000 | US$'000 | US$'000 | US$'000 | US$'000 | US$'000 | US$'000 | |||||||
Cash and cash equivalents | |||||||||||||
Trade receivables | |||||||||||||
Financial assets | |||||||||||||
Trade payables | ( | ( | ( | ( | ( | ( | ( | ||||||
Other current liabilities | ( | ( | |||||||||||
Other non-current liabilities | ( | ||||||||||||
Government grant | ( | ||||||||||||
Decommissioning liability | ( | ||||||||||||
Contingent consideration | ( | ( | |||||||||||
Borrowings | ( | ( | |||||||||||
The balances held at December 31, 2024 that give rise to currency risk exposure are presented in US dollars below:
USD | AUD | EUR | CHF | JPY | GBP | CAD | 1. | |||||||
US$'000 | US$'000 | US$'000 | US$'000 | US$'000 | US$'000 | US$'000 | ||||||||
(Recast) | (Recast) | (Recast) | (Recast) | (Recast) | (Recast) | (Recast) | ||||||||
Cash and cash equivalents | ||||||||||||||
Trade receivables | ||||||||||||||
Financial assets | ||||||||||||||
Trade payables | ( | ( | ( | ( | ( | ( | ( | |||||||
Government grant liability | ( | |||||||||||||
Decommissioning liability | ( | |||||||||||||
Contingent consideration liability | ( | ( | ( | |||||||||||
Borrowings | ( | ( |
F-61
Sensitivity
each reporting date would have on the Group’s reported profit/(loss) after income tax and/or equity balance.
Impact on post-tax profit/(loss)
2025 | 2025 | 2025 | 2025 | 2024 | 2024 | 2024 | 2024 | |||||||||
+10% Profit/ (loss) | -10% Profit/ (loss) | +10% Equity | -10% Equity | +10% Profit/ (loss) | -10% Profit/ (loss) | +10% Equity | -10% Equity | 1. | ||||||||
US$'000 | US$'000 | US$'000 | US$'000 | US$'000 | US$'000 | US$'000 | US$'000 | 1. | ||||||||
(Recast) | (Recast) | (Recast) | (Recast) | 1. | ||||||||||||
AUD | ( | ( | ||||||||||||||
EUR | ( | ( | ( | ( | ||||||||||||
CHF | ( | ( | ||||||||||||||
JPY | ( | ( | ||||||||||||||
GBP | ( | ( | ( | ( | ||||||||||||
CAD | ( | ( | ||||||||||||||
Total | ( | ( | ( | ( |
33.4. Credit risk
Credit risk refers to the risk that a counterparty will default on its contractual obligations resulting in financial loss to the
Group. Credit risk arises from cash and cash equivalents and credit exposures to customers, including outstanding
receivables.
Credit risk is managed on a group basis. If customers are independently rated, these ratings are used. Otherwise, if there
is no independent rating, the Group assesses the credit quality of the customer, taking into account its financial position,
past experience and other factors. Individual risk limits are set based on internal or external ratings. The compliance with
credit limits by customers is regularly monitored. The Group obtains guarantees where appropriate to mitigate credit risk.
The Group applies the IFRS 9 simplified approach to measuring expected credit losses which uses a lifetime expected
loss allowance for all trade receivables.
To measure the expected credit losses, trade receivables have been grouped based on shared credit risk characteristics
and the days past due. The expected loss rates are based on historical payment profiles of sales and the corresponding
historical credit losses experienced. The historical loss rates are adjusted to reflect current and forward-looking
information on macroeconomic factors affecting the ability of the customers to settle the receivables.
Trade receivables are written off where there is no reasonable expectation of recovery. Indicators that there is no
reasonable expectation of recovery include, amongst others, the failure of a debtor to engage in a repayment plan with
the Group, and the failure to make contractual payments for a period of greater than 120 days past due.
Impairment losses on trade receivables are presented within selling, general and administration costs within profit or
loss. Subsequent recoveries of amounts previously written off are credited against the same line item.
As at December 31, 2025, the expected credit losses are $2,377,000 (2024: $131,000 ). The following tables sets out the
aging of trade receivables, according to their due date:
F-62
Aged trade receivables
Expected credit losses | Gross carrying amount | 1. | |||||||
2025 | 2024 | 2025 | 2024 | ||||||
US$'000 | US$'000 | US$'000 | US$'000 | 1. | |||||
(Recast) | (Recast) | 1. | |||||||
Not past due: | |||||||||
Past due: | |||||||||
30 days | ( | ( | |||||||
60 days | ( | ( | |||||||
90 days | ( | ( | |||||||
120 days | ( | ( | |||||||
Total | ( | ( | |||||||
Credit risk concentration profile
The Group has a significant credit risk exposure to three distributors of 71 % (2024: 87 % to three distributors). The Group
defines major credit risk as exposure to a concentration exceeding 10 % of a total class of such asset.
33.5. Liquidity risk
The Group is exposed to liquidity and funding risk from operations and from external borrowings, where the risk is that
the Group may not be able to refinance debt obligations or meet other cash outflow obligations when required. Vigilant
liquidity risk management requires the Group to maintain sufficient liquid assets (mainly cash and cash equivalents). The
Group manages liquidity risk by maintaining adequate cash reserves by continuously monitoring actual and forecast cash
flows and matching the maturity profiles of financial assets and liabilities.
Remaining contractual maturities:
The following tables detail the Group’s remaining contractual maturity for its financial instrument liabilities. The tables
have been drawn up based on the undiscounted cash flows of financial liabilities based on the earliest date on which the
financial liabilities are required to be paid. The tables include both interest and principal cash flows disclosed as
remaining contractual maturities and therefore these totals may differ from their carrying amount in the Consolidated
statement of financial position.
1-6 months | 6-12 months | 1-5 years | Over 5 years | Total contractual cash flows | Carrying amount of liabilities | |||||||
As at December 31, 2025 | US$'000 | US$'000 | US$'000 | US$'000 | US$'000 | US$'000 | ||||||
Non-derivatives | ||||||||||||
Trade and other payables | ||||||||||||
Other non-current liabilities | ||||||||||||
Borrowings | ||||||||||||
Lease liabilities | ||||||||||||
Government grant liability | ||||||||||||
Contingent consideration | ||||||||||||
Total financial liabilities |
F-63
1-6 months | 6-12 months | 1-5 years | Over 5 years | Total contractual cash flows | Carrying amount of liabilities | 1. | ||||||
US$'000 | US$'000 | US$'000 | US$'000 | US$'000 | US$'000 | |||||||
As at December 31, 2024 | (Recast) | (Recast) | (Recast) | (Recast) | (Recast) | (Recast) | ||||||
Non-derivatives | ||||||||||||
Trade and other payables | ||||||||||||
Borrowings | ||||||||||||
Lease liabilities | ||||||||||||
Government grant liability | ||||||||||||
Contingent consideration | ||||||||||||
Total financial liabilities |
This section explains the judgments and estimates made in determining the fair values of the financial instruments that
are recognized and measured at fair value in the financial statements.
33.6.1. Financial assets
Financial assets are categorized as either level 1 or level 3 financial assets and remeasured at each reporting date with
movements recognized in other comprehensive income. The inputs used in the level 1 fair value calculations are with
reference to published price quotations for the associated equity instruments in an active market.
Level 3 financial assets are subject to key assumptions and unobservable inputs which include risk adjusted post-tax
discount rates and forecasted discounted cashflows. These inputs significantly impact the underlying value of these
assets.
Sensitivity of level 1 financial assets
An increase/(decrease) of 10
increase/(decrease) other comprehensive income by $102,000 234,000
Sensitivity of level 3 financial assets
An increase/(decrease) of 10
constant will increase/(decrease) other comprehensive income by $266,000 167,000
33.6.2. Financial liabilities
Contingent consideration liabilities are categorized as level 3 financial liabilities and remeasured at each reporting date
with movements recognized in profit or loss, except in instances where changes are permitted to be added to/reduce an
associated asset. The inputs used in fair value calculations are determined by Management.
The carrying amount of financial liabilities measured at fair value is principally calculated based on inputs other than
quoted prices that are observable for these financial liabilities, either directly (i.e. as unquoted prices) or indirectly (i.e.
derived from prices). Where no price information is available from a quoted market source, alternative market
mechanisms or recent comparable transactions, fair value is estimated based on Management’s views on relevant future
prices, net of valuation allowances to accommodate liquidity, modelling and other risks implicit in such estimates.
Sensitivity of level 3 financial liabilities
The potential effect of using reasonably possible alternative assumptions in valuation models, based on a change in the
most significant input, such as sales volumes, by an increase/(decrease) of 10
will increase/(decrease) profit before tax by $275,000 1,863,000
Valuation processes
The finance team of the Group performs the valuation of contingent consideration liabilities required for financial
reporting purposes, including level 3 fair values. This team reports directly to the Chief Financial Officer ("CFO").
Discussions of valuation processes and results are held between the CFO and Board at least once every six months, in
line with the Group’s half-yearly reporting periods.
F-64
The main level 3 inputs used by the Group in measuring the fair value of contingent consideration liabilities are derived
and evaluated as follows:
•discount rates are determined by an independent third-party using a weighted average cost of capital model to
calculate a post-tax rate that reflects current market assessments of the time value of money and the risk specific
to the asset
•regulatory/marketing authorization approval dates and approval for marketing authorization probability risk factors
are derived in consultation with the Group’s regulatory team
•expected sales volumes and net sales price per unit are estimated based on market information on annual incidence
rates and information for similar products and expected market penetration, and
•contingent consideration cash flows are estimated based on the terms of the sale contract. Changes in fair values
are analyzed at the end of each reporting period during the half-yearly valuation discussion between the CFO and
Board. As part of this discussion the CFO presents a report that explains the reason for the fair value movement.
34. Contingent liabilities
The Group has entered into collaboration arrangements, including in-licensing arrangements with various companies.
Such collaboration agreements may require the Group to make payments on achievement of stages of development,
launch or revenue milestones and may include variable payments that are based on unit sales or profit (e.g. royalty and
profit share payments). The amount of variable payments under the arrangements are inherently uncertain and difficult
to predict, given the direct link to future sales, profit levels and the range of outcomes.
The Group also has certain take or pay arrangements with contract manufacturers or service providers which serve as
commercial manufacturers and suppliers for certain products. To the extent a commitment is determined to be onerous,
these are provided for within provisions in the Consolidated statement of financial position.
The Group has received a subpoena from the U.S. Securities and Exchange Commission ("SEC") seeking various
documents and information primarily relating to the Company’s disclosures regarding the development of the Company’s
prostate cancer therapeutic candidates. This matter remains a fact-finding request. Telix is cooperating with the
subpoena including responding to the SEC’s requests. The SEC has not asserted any charges against the Group or any of
it's personnel, and no conclusions have been reached, or that the SEC has a negative opinion of any person, entity or
security. At this stage, the Group cannot predict the duration, scope or outcome of this matter. The Group will continue
to monitor the inquiry and update its disclosures as appropriate.
On November 10, 2025, a putative securities class action complaint was filed in the United States District Court for the
Southern District of Indiana seeking, among other relief, class certification, designation of a lead plaintiff, damages and a
jury trial. On January 20, 2026, the Group was formally served the complaint on behalf of a purposed U.S. shareholder.
The Group intends to seek dismissal on this matter through early motion practice and will rigorously defend itself. This
case is in its earliest stages; no class has been certified and the court has not ruled on any dispositive motions. At this
stage, the Group cannot predict the duration, scope or outcome of this matter. The Group cannot predict the timing or
outcome of this litigation or estimate a possible loss or range of loss, if any, at this time. The Group will continue to
monitor this matter.
Legal fees and other costs associated with contingent matters, including litigation, regulatory inquiries and
investigations, are expensed as incurred.
35. Commitments
We have commitments against existing development activities and capital commitments relating to the purchase of
isotope raw materials from a vendor over a three year period. R&D commitments are estimated based on the contractual
obligations included within agreements entered into by us, to the extent that a work order has been executed with the
vendor.
Certain of our supply agreements contain minimum purchase commitments in certain situations, the amount and timing of
which are not known. Additionally, we enter into contracts in the normal course of business with clinical trial sites and
clinical supply manufacturers and with vendors for preclinical studies and clinical trials, research supplies and other
services and drugs for operating purposes. These contracts generally provide for termination after a notice period, and,
therefore, are cancellable contracts.
We have entered into collaboration arrangements, including in-licensing arrangements with various companies. Such
collaboration agreements may require us to make payments on achievement of stages of development, launch or
revenue milestones and may include variable payments that are based on unit sales or profit (e.g., royalty and profit
share payments). The amount of variable payments under the arrangements are inherently uncertain and difficult to
predict, given the direct link to future sales, profit levels and the range of outcomes. These payments are not included in
this table of contractual obligations.
F-65
construction of the Brussels South manufacturing facility and the purchase of isotope raw materials from a vendor over a
three year period.
Due < 1 year | Due > 1 year | 1. | ||
US$'000 | US$'000 | 1. | ||
As at December 31, 2025 | ||||
Capital commitments | ||||
R&D commitments | ||||
(Recast) | (Recast) | |||
As at December 31, 2024 | ||||
Capital commitments | ||||
R&D commitments | ||||
36. Related party transactions
36.1. Key management personnel compensation
2025 | 2024 | 2023 | 1. | |||
US$ | US$ | US$ | 1. | |||
(Recast) | (Recast) | 1. | ||||
Short-term employee benefits | ||||||
Superannuation entitlements | ||||||
Share-based payments | ||||||
36.2. Transactions with other related parties
2025 | 2024 | 2023 | ||||
US$ | US$ | US$ | ||||
(Recast) | (Recast) | |||||
Purchases of various goods and services from entities controlled by key management personnel1 | ||||||
1.Non-Executive Director, Dr. Andreas Kluge (previously a non-executive director, retired from the Board on October 17,
2024), is the principal owner and Geschäftsführer (Managing Director) of ABX-CRO, a clinical research organization
(CRO) that specializes in radiopharmaceutical product development. Following retirement as a Non-Executive
Director, Dr. Kluge has been engaged by Telix on a consultancy basis and will continue to provide the Board of
Directors strategic advice alongside clinical input into key development programs, reflective of his ongoing
Transactions with Andreas Kluge and ABX-CRO do not meet the criteria of a related party transaction following Andreas
Kluge's retirement as a non-executive director on October 17, 2024.
In March 2024, the Group entered into an agreement to purchase the QDOSE dosimetry software platform from ABX-
CRO. QDOSE is a software platform designed to enable reliable estimation of patient-specific dosimetry for both
therapeutic and diagnostic radiopharmaceuticals. We agreed to pay ABX-CRO upfront cash consideration of €1,200,000 ,
a share of profits generated from QDOSE sales and a referral fee on deals referred from or initiated by ABX-CRO over a
During the year ended December 31, 2024 the total amount paid was $513,809 , (2023: $835,315 ) and the amount
payable to ABX-CRO at December 31, 2024 was $nil (2023: $nil ) respectively.
F-66
Transactions with related parties are undertaken on an arm’s length basis and competitive with quotes obtained from
other suppliers for similar services.
36.3. Interests in other entities
capital consisting solely of ordinary shares that are held directly by the Group, and the proportion of ownership interests
held equals the voting rights held by the Group. The country of incorporation or registration is also the principal place of
business.
Name of entity | Country of incorporation | Ownership interest held by the Group (%) |
Telix Pharmaceuticals Ltd | Australia | N/A |
Telix Pharmaceuticals (Innovations) Pty Ltd1 | Australia | |
Telix Pharmaceuticals Holdings Pty Limited1 | Australia | |
Telix Pharmaceuticals International Holdings Pty Ltd1 | Australia | |
Telix Pharmaceuticals Australia Holdings Pty Ltd1 | Australia | |
Telix Pharmaceuticals (ANZ) Pty Ltd1 | Australia | |
Telix Pharmaceuticals (Corporate) Pty Ltd1 | Australia | |
Telix Pharmaceuticals (Belgium) SRL | Belgium | |
Telix Innovations SA | Belgium | |
Telix Innovations Rph Participacoes Ltda | Brazil | |
Telix Pharmaceuticals (Canada) Inc. | Canada | |
Telix ARTMS Inc. | Canada | |
Telix Pharmaceuticals (France) SAS | France | |
Telix Pharmaceuticals (Germany) GmbH | Germany | |
Rhine Pharma GmbH2 | Germany | |
Therapeia GmbH & Co. KG | Germany | |
Therapeia Verwaltungs-GmbH | Germany | |
Telix Pharma Japan KK | Japan | |
Telix Pharmaceuticals (NZ) Limited | New Zealand | |
Telix Pharmaceuticals (Singapore) Pte Ltd | Singapore | |
Telix Pharmaceuticals (Switzerland) GmbH | Switzerland | |
Telix Pharmaceuticals (UK) Ltd | United Kingdom | |
Lightpoint Surgical Ltd | United Kingdom | |
Lightpoint Surgical Spain S.L. (Lightpoint Medical Espana SLU) | Spain | |
Telix Pharmaceuticals (US) Inc. | USA | |
Telix Optimal Tracers, LLC | USA | |
Telix IsoTherapeutics Group, Inc. | USA | |
Telix QSAM, Inc. | USA | |
QSAM Therapeutics Inc. | USA | |
RLS (USA), Inc. | USA | |
Las Vegas Radiopharmacy, Inc. | USA | |
Telix Targeting Technologies, Inc. | USA | |
ARTMS US, Inc. | USA |
1.Denotes an entity that is a party to a deed of cross guarantee, refer to note 39 for further information.
F-67
2.The Group plans to spin off this entity and has granted options to certain third parties to acquire an economic interest
in the entity once key milestones are achieved.
37. Remuneration of auditor
Auditors of the Group - PricewaterhouseCoopers Australia and related network firms | 2025 | 2024 | 2023 | 1. | ||
US$ | US$ | US$ | 1. | |||
(Recast) | (Recast) | 1. | ||||
Audit or review of financial statements | ||||||
Other assurance services | ||||||
Other advisory services | ||||||
Other assurance services relate to assurance and associated services that are traditionally performed by an independent
auditor, including accounting consultation and consultation concerning financial accounting, reporting standards and due
diligence. Other assurance services in 2024 includes comfort letter assurance services performed as part of the Group's
U.S. listing.
Other auditors and their related network firms | 2025 | 2024 | 2023 | 1. | ||
US$ | US$ | US$ | 1. | |||
(Recast) | (Recast) | 1. | ||||
Audit or review of financial statements | ||||||
Other advisory services | ||||||
38. Parent entity financial information
Australian Disclosure Requirements
The financial information for the parent entity has been prepared on the same basis as the consolidated financial
statements. The individual financial statements for the parent entity show the following aggregate amounts:
F-68
Statement of financial position | 2025 | 2024 | |
$’000 | $’000 | ||
(Recast) | |||
Assets | |||
Current assets | |||
Non-current assets | |||
Total assets | |||
Liabilities | |||
Current liabilities | |||
Non-current liabilities | |||
Total liabilities | |||
Net assets | |||
Equity | |||
Share capital | |||
Share capital reserve | ( | ||
Other reserves | |||
Retained earnings/(accumulated losses) | ( | ||
Total equity | |||
Statement of comprehensive income | |||
Loss for the year | ( | ( | |
Total comprehensive loss for the year | ( | ( |
39. Deed of cross guarantee
Australian Disclosure Requirements
The Company and certain Australian subsidiaries of the Group have entered into a deed of cross guarantee. By entering
into the deed, the subsidiaries which are party to the deed have been relieved from the requirement to prepare and
lodge audited financial statements under ASIC Corporations (Wholly-owned Companies) Instrument 2016/785. The
subsidiaries identified with a ‘1’ in note 35.3 are parties to a deed of cross guarantee under which each Company
guarantees to each creditor payment in full of any debt in accordance with the deed of cross guarantee.
For the year ended December 31, 2025 the parties to the deed of cross guarantee generated a loss of $99,349,000
(2024: profit of $16,291,000 ) and as at December 31, 2025 were in an asset position of $176,579,000 (2024: net asset
position $189,036,000 ), with cash and cash equivalents of $84,572,000 (2024: $326,514,000 ).
Cash on hand and the repatriation of future cash inflows from commercial activities undertaken by wholly-owned foreign
subsidiaries is considered sufficient to meet forecast cash outflows, research and development activities currently
underway and other committed business activities for at least 12 months from the date of these financial statements.
Further, current liabilities include loans with other subsidiaries in the Group of $47,190,000 (2024: $29,965,953 ) which
will be settled when sufficient funds are available.
On this basis, the Directors are satisfied that the parties to the deed of cross guarantee continue to be a going concern
as at the date of these financial statements.
deed of cross guarantee are provided as follows:
F-69
Consolidated statement of comprehensive income or loss | 2025 | 2024 | |
US$'000 | US$'000 | ||
(Recast) | |||
Revenue from contracts with customers | |||
Cost of sales | ( | ( | |
Gross profit | |||
Research and development costs | ( | ( | |
Selling and marketing expenses | ( | ( | |
Manufacturing and distribution costs | ( | ( | |
General and administration costs | ( | ( | |
Other (losses)/gains (net) | ( | ||
Operating profit/(loss) | ( | ||
Finance income | |||
Finance costs | ( | ( | |
(Loss)/profit before income tax | ( | ||
Income tax expense | ( | ( | |
(Loss)/profit from continuing operations after income tax | ( | ||
Changes in the fair value of equity investments at fair value through other comprehensive income | ( | ( | |
Total comprehensive (loss)/income for the year | ( |
F-70
Consolidated statement of financial position | 2025 | 2024 | |
US$'000 | US$'000 | ||
(Recast) | |||
Current assets | |||
Cash and cash equivalents | |||
Trade and other receivables | |||
Inventories | |||
Other current assets | |||
Total current assets | |||
Non-current assets | |||
Net investment in subsidiaries | |||
Intangible assets | |||
Property, plant and equipment | |||
Right-of-use assets | |||
Financial assets | |||
Deferred tax assets | |||
Other non-current assets | |||
Total non-current assets | |||
Total assets | |||
Current liabilities | |||
Trade and other payables | |||
Contract liabilities | |||
Lease liabilities | |||
Borrowings | |||
Contingent consideration | |||
Employee benefit obligations | |||
Total current liabilities | |||
Non-current liabilities | |||
Contract liabilities | |||
Lease liabilities | |||
Borrowings | |||
Contingent consideration | |||
Deferred tax liabilities | |||
Employee benefit obligations | |||
Total non-current liabilities | |||
Total liabilities | |||
Net assets | |||
Equity | |||
Share capital | |||
Share capital reserve | ( | ||
Other reserve | |||
Accumulated losses | ( | ( | |
Total equity |
F-71
40. Events occurring after the reporting period
the year ended December 31, 2025.
1 All entities are corporate entities.
F-72
Australian Disclosure Requirements
Consolidated entity disclosure statement
Basis of preparation
This consolidated entity disclosure statement ("CEDS") has been prepared in accordance with the Australian
Corporations Act 2001 (Cth) and includes information for each entity that was part of the consolidated entity as at the
end of the financial year in accordance with AASB 10 Consolidated Financial Statements.
Name of entity1 | Tax residency | |||
Country of incorporation | Ownership interest held by the Group (%) | Australian or foreign | Foreign jurisdiction(s) | |
Telix Pharmaceuticals Ltd | Australia | N/A | Australian | N/A |
Telix Pharmaceuticals (Innovations) Pty Ltd | Australia | 100 | Australian | N/A |
Telix Pharmaceuticals Holdings Pty Limited | Australia | 100 | Australian | N/A |
Telix Pharmaceuticals International Holdings Pty Ltd | Australia | 100 | Australian | N/A |
Telix Pharmaceuticals Australia Holdings Pty Ltd | Australia | 100 | Australian | N/A |
Telix Pharmaceuticals (ANZ) Pty Ltd | Australia | 100 | Australian | N/A |
Telix Pharmaceuticals (Corporate) Pty Ltd | Australia | 100 | Australian | N/A |
Telix Pharmaceuticals (Belgium) SRL | Belgium | 100 | Foreign | Belgium |
Telix Innovations SA | Belgium | 100 | Foreign | Belgium |
Telix Innovations Rph Participacoes Ltda | Brazil | 51 | Foreign | Brazil |
Telix Pharmaceuticals (Canada) Inc. | Canada | 100 | Foreign | Canada |
Telix ARTMS Inc. | Canada | 100 | Foreign | Canada |
Telix Pharmaceuticals (France) SAS | France | 100 | Foreign | France |
Telix Pharmaceuticals (Germany) GmbH | Germany | 100 | Foreign | Germany |
Rhine Pharma GmbH3 | Germany | 100 | Foreign | Germany |
Therapeia GmbH & Co. KG | Germany | 100 | Foreign | Germany |
Therapeia Verwaltungs-GmbH | Germany | 100 | Foreign | Germany |
Telix Pharma Japan KK | Japan | 100 | Foreign | Japan |
Telix Pharmaceuticals (NZ) Limited | New Zealand | 100 | Australian | New Zealand |
Telix Pharmaceuticals (Singapore) Pte Ltd | Singapore | 100 | Australian | Singapore |
Telix Pharmaceuticals (Switzerland) GmbH | Switzerland | 100 | Foreign | Switzerland |
Telix Pharmaceuticals (UK) Ltd | United Kingdom | 100 | Australian | United Kingdom |
Lightpoint Surgical Ltd | United Kingdom | 100 | Foreign | United Kingdom |
Lightpoint Surgical Spain S.L. (Lightpoint Medical Espana SLU) | Spain | 100 | Foreign | Spain |
Telix Pharmaceuticals (US) Inc. | USA | 100 | Foreign | USA |
Telix Optimal Tracers, LLC | USA | 100 | Foreign | USA |
Telix IsoTherapeutics Group, Inc. | USA | 100 | Foreign | USA |
Telix QSAM, Inc. | USA | 100 | Foreign | USA |
QSAM Therapeutics Inc. | USA | 100 | Foreign | USA |
RLS (USA), Inc. | USA | 100 | Foreign | USA |
F-73
Name of entity1 | Tax residency | |||
Country of incorporation | Ownership interest held by the Group (%) | Australian or foreign | Foreign jurisdiction(s) | |
Las Vegas Radiopharmacy, Inc. | USA | 100 | Foreign | USA |
Telix Targeting Technologies, Inc. | USA | 100 | Foreign | USA |
ARTMS US, Inc. | USA | 100 | Foreign | USA |
Australian Disclosure Requirements
Determination of tax residency
Section 295 (3A)(vi) of the Australian Corporations Act 2001 (Cth) defines tax residency as having the meaning in the
Income Tax Assessment Act 1997. The determination of tax residency involves judgment as it can be fact dependent and
subject to interpretation, requiring consideration of matters such as location of central management and control or place
of effective management.
The rules and guidance in respect of tax residency have been applied in good faith. In determining tax residency, the
consolidated entity has applied the following interpretations:
•Australian tax residency: The consolidated entity has applied current legislation and judicial precedent, including
having regard to the Tax Commissioner’s public guidance in Tax Ruling TR 2018/5.
•Foreign tax residency: The consolidated entity has applied current legislation, judicial precedent and practice in
the determination of foreign tax residency.
F-74
Directors’ declaration
1.In the opinion of the Directors:
a.the financial statements and notes of the Company and Group are in accordance with the Australian Corporations
Act 2001 (Cth), including:
i.complying with applicable Accounting Standards, the Corporations Regulation 2001 and other mandatory
professional reporting requirements; and
ii.giving a true and fair view of the Company's and Group's financial position as at December 31, 2025 and of
their performance for the year ended on that date.
b.there are reasonable grounds to believe that the Company will be able to pay its debts as and when they become
due and payable.
2.Within the notes to the financial statements it is confirmed that the financial statements also comply with the
International Financial Reporting Standards as issued by the International Accounting Standards Board and as
disclosed in note 2.2.
3.In the opinion of the Directors, as at the date of this declaration, there are reasonable grounds to believe that the
Company and entities identified in note 39 will be able to meet any obligations or liabilities to which they are or may
become subject by virtue of the Deed of Cross Guarantee between the Company and those entities pursuant to ASIC
Corporations (Wholly-Owned Companies) Instrument 2016/785.
4.In the opinion of the Directors the consolidated entity disclosure statement required by subsection 295(3A) of the
Australian Corporations Act 2001 (Cth), as disclosed in the Consolidated entity disclosure statement on pages F-72 to
F-73, is true and correct.
5.This declaration has been made after receiving the declarations required to be made to the Directors in accordance
with section 295A of the Australian Corporations Act 2001 (Cth) for the financial year ended December 31, 2025.
6.Signed in accordance with a resolution of the Directors.
Mark Nelson | Christian Behrenbruch |
Interim Chair | Managing Director and Group CEO |
February 20, 2026 | February 20, 2026 |
Exhibit 4.3

Deed of Indemnity, Access and Insurance
Telix Pharmaceuticals Limited
ACN 616 620 369
and
INSERT FULL NAME OF DIRECTOR
Page | 2
Table of Contents |
to update table of contents - press F9
Page | 3
Page | 4
Deed of Indemnity |
This deed is made on [insert date] between the following parties:
Telix Pharmaceuticals Limited ACN 616 620 369 of 55 Flemington Road, North Melbourne VIC 3051 Australia (the Company); and |
[insert full name of Director] of [insert Director address] (the Director). |
Background | The Constitution provides that the Company: •must indemnify directors to the full extent permitted by law; and •may enter into contracts of insurance to protect directors against any liability incurred by directors as directors of the Company. | ||
(A) | The Director has been a director of the Company or Relevant Company (as the case may be) as of the Appointment Date. | ||
(B) | In consideration of the Director agreeing to act as a director of the Company or Relevant Company (as the case may be), the Company agrees to: | ||
(i) | indemnify the Director against Liabilities incurred while acting as officer of the Company or Relevant Company (as the case may be); and | ||
(ii) | maintain a D&O Policy, | ||
(iii) | provide access to Board Papers, | ||
on the terms contained in this deed. | |||
This deed witnesses that in consideration of, among other things, the mutual promises contained in
this deed, the parties agree as follows:
1Definitions and Interpretation |
1.1Definitions
In this deed:
Defined term | Meaning |
Affiliate | has the meaning as defined under the 15 U.S Code. |
Appointment Date | the date the Director was appointed a director of the Company or as a director of Relevant Company (as the case may be). |
Board | the board of directors of the Company. |
Page | 5
Defined term | Meaning |
Board Papers | in relation to the Company: 1all material provided to the Director, provided to or tabled at any meeting of the Board, or to any committee of the Board (Material), whether in hard copy or electronic form, including without limitation board papers, committee papers, correspondence, submissions, minutes, legal advice, reports and financial statements; and 1all documents of the Company or to which the Company is a party where those documents are referred to in any Material, during the Relevant Period. and, in relation to each Relevant Company: 2all material provided to the Director in his/her capacity as a director of the Relevant Company, provided to or tabled at any meeting of the board of the Relevant Company or to any committee of the board of the Relevant Company, whether in hard copy or electronic form, including without limitation board papers, committee papers, correspondence, submissions, minutes, legal advice, reports, and financial statements; and 3all documents of the Relevant Company or to which the Relevant Company is a party referred to in any such material. |
Business Day | a day other than a Saturday, Sunday, bank holiday or public holiday in Melbourne, Victoria. |
Claim | (a)any legal proceeding, administrative proceeding, arbitral proceeding, investigation or enquiry, mediation, or other form of alternative dispute resolution, arising out of or in connection with any act or omission by the Director or otherwise involving the Director in their capacity as a director; and (b)any written or oral threat, complaint or demand or other circumstances that might reasonably lead to the Director considering that any proceedings set out in paragraph (a) will be commenced. |
Constitution | the Company’s constitution as amended, varied or replaced from time to time. |
Corporations Act | the Corporations Act 2001 (Cth). |
D&O Policy | a policy of insurance insuring the Director (amongst others) against liability in their capacity as a director and officer of the Company and its Related Bodies Corporate. |
director | has the same meaning given in section 9 of the Corporations Act. |
Page | 6
Defined term | Meaning |
document | has the meaning given to it for the purposes of the Acts Interpretation Act 1901 (Cth). |
Group Entities | the Company and any Subsidiary or Affiliate of the Company. |
Liability | a liability of any kind (whether actual or contingent and whether fixed or ascertained) including costs, damages, fees, expenses, and including whether the costs and expenses are incurred in connection with any investigation or inquiry by a government agency or liquidator. |
officer | has the meaning given to it for the purposes of the Corporations Act. |
Permitted Purpose | (a)defending or responding to an action or proceeding (or preparing to defend an action or proceeding which the Director has reason to believe will be brought against them) which relates to an act or omission of the Director in providing services in their capacity as an officer of the Company or Relevant Company (as the case may be) during the Relevant Period; (b)appearing before an inquiry or hearing of a Regulatory Body (or preparing for an inquiry or hearing of a Regulatory Body) where the Director has reason to believe that the Director will be required to appear before that inquiry or hearing relating to an act or omission of the Director in providing services in their capacity as a director of the Company or Relevant Company (as the case may be) during the Relevant Period; (c)conducting or preparing to conduct an action or proceeding which the Director in good faith proposes to bring relating to an act or omission of the Director in providing services in their capacity as officer of the Company or Relevant Company (as the case may be) during the Relevant Period; or (d)disclosing Board Papers to third parties (including, without limitation, the Director’s legal and other professional advisors) where such disclosure is necessary in relation to a matter under any of sub- clauses (a), (b) or (c) of this clause; or (e)any other purpose which the Company has provided written consent. |
Page | 7
Defined term | Meaning |
Proceedings | (a)any investigation, hearing, inquiry or review undertaken by a court, arbitrator, mediator or tribunal, governmental, administrative or Regulatory Body, or public authority; and (b)any procedural step relating to such a hearing, conference, dispute, inquiry or investigation, under or in respect of which the Director is being examined or is involved because the Director is or was a director of the Company or a Relevant Company (as the case may be) in the Relevant Period. |
Protection Period | in relation to the Company and each Relevant Company (as the case may be) the period commencing on the Appointment Date or the date of this deed (whichever is earlier) and ending on the later of: (a)the date which is 7 years after the Director ceases to hold office as a director of the Company or the Relevant Company (as the case may be); and (b)the date any Proceedings commenced during the period specified in paragraph (a) have been finally resolved. |
Regulatory Body | an entity constituted under the laws of Australia or any other jurisdiction which has the power to regulate the conduct and affairs of a Group Entity and shall include (without limitation) the Australian Securities and Investment Commission, the Australian Competition and Consumer Commission and the Australian Tax Office. |
Related Body Corporate | has the meaning given to it in section 50 of the Corporations Act, and includes a Subsidiary and an Affiliate. |
Relevant Company | each Related Body Corporate of the Company of which the Director is a director from time to time. |
Relevant Period | the period commencing on the Appointment Date and ending on the date the Director ceases to act as a director of the Company or the Relevant Company (as the case may be). |
Subsidiary | has the meaning given in section 9 of the Corporations Act and refers to any corporation which before, at or after the date of this deed was, is or becomes a Subsidiary of the Company. |
1.2Interpretation
In this deed, unless the context requires otherwise:
(a)terms used from the Corporations Act have the meaning given under section 9 of the
Corporations Act;
Page | 8
(b)words importing the singular include the plural and vice versa;
(c)words importing a gender include any gender;
(d)a reference to a thing (including, but not limited to, a right) includes any part of that
thing;
(e)a reference to a right includes a remedy, power, authority, discretion or benefit;
(f)other parts of speech and grammatical forms of a word or phrase defined in this deed
have a corresponding meaning;
(g)a reference to a clause or party is a reference to a clause of, and a party to, this deed;
(h)a reference to a document or deed includes all amendments or supplements to, or
replacements or novations of, that document or deed;
(i)a reference to the Director includes the Director’s personal representatives;
(j)a reference to a statute, legislation, regulation, proclamation, ordinance or by-law
includes all statutes, regulations, proclamations, ordinances or by-laws amending,
consolidating or replacing it, and a reference to a statute includes all regulations,
proclamations, ordinances and by-laws issued under that statute;
(k)a reference to a party to a document includes that party’s successors and permitted
assignees;
(l)a rule of construction does not apply to the disadvantage of a party because that
party was responsible for the preparation of this deed or any part of it; and
(m)if a day for payment upon which an obligation must be performed is not a Business
Day the payment is due on the next Business Day.
2Indemnity |
1.1General indemnity
(a)To the maximum extent permitted by law, the Company indemnifies the Director on a
full indemnity basis, with effect from the Appointment Date and on the terms set out in
this deed, against:
(i)all Liabilities incurred by the Director as an officer of the Company or of a
Related Body Corporate;
(ii)all Liabilities incurred by the Director in relation to actual, threatened or
potential Proceedings; and
(iii)subject to clause 2.1(b), all Liabilities incurred by the Director in the Director’s
capacity as an officer of the Company or of a Related Body Corporate for
Proceedings brought by the Director against third parties in order to protect
the Director’s interests or reputation. This indemnity includes, without
limitation, a liability for reasonable legal costs on a solicitor and own-client
basis.
(b)The Company will only indemnify the Director under clause 2.1(a)(iii) if the Director
first obtains the written consent of the Board which:
(i)must not be unreasonably withheld or delayed; and
(ii)may be provided subject to any conditions the Board considers appropriate
(acting reasonably) including, without limitation, as to the treatment of all or
any damages or other compensation received by the Director in respect of
any Proceedings.
1.2Continuing indemnity
(a)The indemnity in clause 2.1 is an irrevocable, unconditional, continuing and principal
obligation of the Company despite:
Page | 9
(i)the resignation or removal of the Director as a director of the Company;
(ii)the settlement of any dispute between the Director and the Company or a
third party;
(iii)any amendment or variation to, or replacement of, the Constitution;
(iv)any intermediate payments, settlement of accounts or payments;
(v)laches, acquiescence or delay on the part of the Director;
(vi)the death, bankruptcy, insolvency or liquidation of any person or corporation;
or
(vii)the occurrence of any other thing, including any other thing which might
otherwise affect the indemnity whether at law or in equity,
and remains in full force and effect until released by the Director.
(b)The Company shall not be obliged to indemnify the Director under this deed where
the Director fails to perform any of the obligations set out in clause 3.5 to the material
prejudice of the Company.
(c)The indemnity in clause 2.1 enures to the benefit of the Director’s estate.
1.3Additional indemnity
The indemnity in clause 2.1 is in addition to any indemnity contained in the Constitution from
time to time.
3Conduct and obligations |
1.1Notification
The Director must give notice to the Company as soon as reasonably practicable after the
Director becomes aware of any facts, matters, circumstances, or any threatened or pending
Claim against the Director or decision to make a Claim against a third party, which could give
rise to a Claim for indemnification under this deed.
1.2Advancement or payment of costs
(a)On the Director’s request, the Company will advance to or pay on behalf of the
Director, reasonable costs incurred or expected to be incurred by the Director
(whether legal or otherwise) in connection with Proceedings on terms determined by
the Board except that any such advance will be interest free and on an unsecured
basis.
(b)The Director must furnish the Company with invoices or other relevant evidence
satisfactory to the Company of the costs incurred or expected to be incurred for the
purposes of clause 3.2(a).
(c)If the Company has advanced an amount for costs under clause 3.2(a), the amount of
the advance will be in part satisfaction of the Company’s obligation to indemnify the
Director and will cease to be repayable unless it is subsequently found that the
Director was not entitled to be indemnified for those costs.
(d)If the Director has a right of recovery from a third party in respect of some or all of the
Liabilities, the Director must use best efforts to exercise the right of recovery within 10
days of payment being received and account to the Company for any such amount.
(e)If the Company has advanced or otherwise paid an amount under clause 3.2(a) and it
is subsequently found that the Director was not entitled to the advancement or
payment of those costs, the Director must repay this amount to the Company within
30 days of a request from the Company being received by the Director.
Page | 10
1.3Proceedings
(a)The Company may:
(i)assume the conduct, negotiation or defence of a Claim;
(ii)institute legal proceedings (including a counterclaim or cross claim) in relation
to a Claim; and
(iii)subject to clause 3.4, retain lawyers to act on behalf of both the Company
and the Director in relation to the Claim,
(iv)and, when it does so, the conduct of the claim will be under the management
and control of the Company or its insurers (acting reasonably).
(b)The Company must:
(i)notify the Director as soon as reasonably practicable if it intends to take any
action under clause 3.3(a);
(ii)consider the reputation of the Director in acting under clause 3.3(a); and
(iii)not unreasonably withhold or delay its decision under clause 3.3(b)(ii).
(c)If the Company does not elect to take control of the conduct of proceedings under
clause 3.3(a), the Director must ensure that the Company is kept fully informed of any
actual or proposed developments (including, without limitation, any meetings) and is
provided with copies of all material correspondence and documentation relating to
such third party Claim or action, and such other information, assistance and access to
records and personnel as the Company reasonably requires, other than any
correspondence, documents or information to which any legal professional privilege
attaches for the benefit solely of the Director.
1.4Legal representation
(a)The Director may obtain independent legal representation in respect of the conduct,
negotiation or defence of any advice, Claim or Proceeding against the Director as a
result of or arising from being a director of the Company or a Related Body
Corporate.
(b)The Company will reimburse the Director within 10 days of receipt of a written request
for reimbursement from the Director for expenses payable by the Company under this
deed to the extent that those expenses are:
(i)reasonable and incurred prior to the Company assuming conduct of the
Claim;
(ii)incurred with the Company’s prior written authority (which must not be
unreasonably withheld or delayed); and
(iii)reasonable and incurred where it could be reasonably expected that a conflict
between the interests of the Director and the Company (or a Related Body
Corporate) would arise should the same lawyers act on behalf of both parties.
1.5Director’s obligations
To the extent permitted by law, the Director must:
(a)give notice to the Company promptly on becoming aware of any claim, or any
circumstances which could reasonably be expected to give rise to any claim, that may
give rise to a right for the Director to be indemnified under this deed;
(b)take any reasonable action to avoid, resist, dispute, bring an appeal in, compromise
or defend any Claim;
(c)not make an admission of liability or payment in respect of or settle or compromise
any Claim without the Company’s prior written consent (which must not be
unreasonably withheld or delayed);
Page | 11
(d)render all reasonable assistance to the Company in the conduct of any Claim,
including (but not limited to) providing documents, authorities and directions that the
Company requires to prosecute or advance any cross claim or counterclaim; and
(e)on request by the Company, do anything reasonably necessary or desirable to enable
the Company (so far as possible) to be subrogated to and enjoy the benefits of the
Director’s rights in relation to any cross-claims or any Claims against any third party
and render any assistance that is reasonably requested by the Company for the
purpose,
and the Company is not obliged to indemnify the Director under this deed or otherwise where
the Director fails to perform any of these obligations to the material prejudice of the Company.
4Insurance |
1.1Obligation to maintain D&O Policy
(a)Subject to clause 4.2 and to the extent permitted by law, the Company must:
(i)at all times during the Protection Period maintain a D&O Policy consistent
with clause 4.2; and
(ii)not undertake any actions which will, or is likely to, reduce or invalidate the
cover under the D&O Policy, other than making a claim.
1.2Terms and conditions of Policy
(a)Subject to law, the terms of the D&O Policy must, to the extent that such a policy is
available from a reputable insurance company at reasonable commercial rates:
(i)cover the Liabilities which may be incurred by the Director during the
Protection Period arising from the Director acting as a director of the
Company; and
(ii)provide for the terms, conditions, exclusions and additional cover (including
premiums, insuring clauses, exclusions and excess amounts) as are
reasonably appropriate for a company of similar industry and annual revenue;
and
(iii)be in a form and extend to an amount of cover substantially similar to the
policy existing at the time of entering into the deed and in any event is at least
as comprehensive as the insurance policies effected for the benefit of the
existing directors of the Company from time to time during the Protection
Period.
1.3Obligations of Director
(a)The Director undertakes to comply with the obligations and requirements provided for
under the D&O Policy and to take reasonable steps to enable the Company to
maintain the D&O Policy under this deed.
(b)The Director undertakes to disclose to the Company all known relevant matters which
may lead to a claim being made against the Company or the Director, as soon as
practicable after becoming aware of such matters.
(c)To the extent required to comply with its disclosure obligations in relation to the D&O
Policy, the Company will seek information from the Director within a reasonable
period before the renewal of the policy.
(d)The Director acknowledges that negotiations undertaken by the Company on the
terms of the D&O Policy may result in the insurer varying the terms of the D&O Policy.
1.4Obligations of Company
(a)The Company must provide the Director with a copy of the D&O Policy and certificate
of insurance upon request by the Director.
(b)The Company must:
Page | 12
(i)to the maximum extent permitted by law, pay the cost of any premiums under
the D&O Policy;
(ii)not do any act which will, or is likely to, reduce the cover provided for under
the D&O Policy (other than by making a claim under the D&O Policy); and
(iii)notify the Director upon becoming aware that the D&O Policy has been
cancelled, not renewed, or there is a material reduction in the terms of the
D&O Policy.
5Access to Board Papers |
1.1Company to maintain records
(a)Subject to clause 5.1(b), the Company must maintain, and must use all reasonable
endeavours to procure that each Relevant Company maintains, a complete set of
Board Papers in an organised and secure manner for the duration of the Protection
Period.
(b)Where Board Papers were bought into existence before the date of this deed, the
Company complies with its obligations under clause 5.1(a) if it uses all reasonable
endeavours to collate and keep, and if it uses all reasonable endeavours to procure
each Relevant Company to collate and keep, those Board Papers in the manner
required by clause 5.1(a).
1.2Right of inspection
(a)Subject clause 5.4, where a request is received during the Protection Period in
accordance with clause 5.3, the Company must within 7 Business Days, permit the
Director (or a person nominated in writing by the Director) to inspect (at the
Company’s registered office or any other place agreed by the Company and the
Director) and copy Board Papers during Business Hours at no cost to the Director,
where such request is in relation to:
(i)Proceedings brought or which may be brought against the Director, or to
which the Director is a party; or
(ii)a Permitted Purpose.
(a)Where the request relates to a Board Paper of a Relevant Company, the Company
must use all reasonable endeavours to procure the Relevant Company to take the
steps contemplated by clause 5(a).
1.3Access request
Any request for access to, or for a copy of, a Board Paper during the Protection Period must
be made by the Director to the Company in writing, and include particulars of the Board
Papers which are the subject of the request and the purpose for which access is required.
1.4Ownership of Board Papers
The Director acknowledges that the Company or the Relevant Company (as the case may be)
remains the owner of all Board Papers and may request the Director to return or destroy all
copies of Board Papers within 7 Business Days after such time as the purpose for the access
request made under clause 5.3 no longer exists.
1.5Privilege
(a)If the Company or Relevant Company (as the case may be) has any right (including a
right it has jointly or in common with the Directors and others) to privilege, such as
legal professional privilege, in respect of any document which the Director inspects,
copies or uses under this deed or under the Corporations Act or under the general
law rights of a director:
(i)that document is to be taken to be confidential;
(ii)by permitting the inspection, copying or use to the Director or the Director’s
permitted nominee, the Company does not waive any privilege; and
Page | 13
(iii)in so inspecting, copying or using the document by himself or herself or
through the Director’s permitted nominee, the Director must use best efforts
to ensure that so far as is practical the right to privilege is not lost or waived,
whether by the Director or the Director’s nominee or otherwise.
(b)Nothing in this deed or done pursuant to this deed prevents the Company or Relevant
Company (as the case may be) from relying on privilege in proceedings between the
Director and the Company or Relevant Company (including in respect of a document
which the Company or Relevant Company has disclosed to the Director outside those
proceedings).
1.6No limitation
This clause 5 does not limit any right of access the Director otherwise has to Board Papers.
6Confidentiality and privilege |
(a)Subject to any obligation under law, the Director must during the term of office and
after ceasing to be a director, maintain the confidentiality of all information contained
in a Board Paper unless:
(i)disclosure is required by law;
(ii)the Company or the Relevant Company (as the case may be) has given its
prior written consent to disclosure;
(iii)the disclosure is made for the purpose of obtaining professional advice or for
a proper purpose and in the due performance of the Director’s duties; or
(iv)the information is required for the purpose of Proceedings or the threat of
Proceedings in relation to which the Director was given access to the Board
Papers,
and the Director uses the Director’s best endeavours to ensure all information
disclosed is kept confidential.
(b)Nothing in clause 6(a) is intended to replace or reduce the duties which the Director
may have, at law, to the Company or a related body corporate.
(c)Where the Director is entitled to disclose confidential information under clause 6(a)
and the information is that to which legal professional privilege attaches:
(i)unless the Company or the Relevant Company (as the case may be)
expressly states otherwise, it is the Company’s or the Relevant Company’s
(as the case may be) intention that the giving of access to the Director in no
way waives or diminishes the Company’s or the Relevant Company’s right to
claim legal professional privilege; and
(ii)the Director must use its best endeavours to avoid doing anything which will
cause privilege to be waived, extinguished or lost by the Company or the
Relevant Company in relation to third parties.
7Taxation and costs |
1.1Taxation and duty
(a)If a government authority imposes any tax on a sum paid to the Director under this
deed, then the Company or the Relevant Company (as the case may be) must
increase the amount paid to the Director so that the net amount to which the Director
becomes entitled after deduction of all applicable taxes is equal to that which would
have been payable under the deed had no such tax been imposed.
(b)The Company or the Relevant Company (as the case may be) must pay any stamp
duty chargeable on this deed.
1.2Costs
Each party must bear its own costs of negotiating, preparing and executing this deed.
Page | 14
8General |
1.1Governing law and jurisdiction
This deed is governed by the laws of Victoria, Australia and each party irrevocably submits to
the non-exclusive jurisdiction of the courts of Victoria, Australia.
1.2Counterparts
This deed may be executed in counterparts. All executed counterparts constitute one
document.
1.3Unenforceable provision
If a provision in this deed is wholly or partly invalid or unenforceable in any jurisdiction, the
provision or part of it that is invalid or unenforceable must, to that extent and in that
jurisdiction, be treated as deleted from this deed. Any provision removed under this clause 8.3
does not affect the validity or enforceability of the remaining provisions in that jurisdiction or
any other jurisdiction.
1.4Survival
Each obligation of confidence and indemnity in this deed is a continuing obligation, separate
and independent from the other obligations and survives termination of this deed for any
reason.
1.5Further action
Each party must, at its own expense, do all things reasonably necessary (including executing
all documents) to give full effect to this deed and the transactions contemplated by it.
1.6Exclusion of moratoria
Any statute, moratorium or other governmental order that prejudicially affects the rights,
powers or discretions of the parties under this deed does not apply to this deed unless the
application is mandatory.
1.7Variation and waiver
(a)This deed may only be varied in writing signed by each party.
(b)A party does not:
(i)waive a right, power, or remedy unless it does so in writing and any such
waiver is only effective for the specific instance for which it is given; or
(ii)waive a right, power, or remedy if it fails to exercise or delays in exercising
the right, power or remedy.
(c)A single or partial exercise of a right by a party does not prevent another further
exercise of that right, power, or remedy.
1.8Notices
Any notices or other communications under this deed must be in writing addressed to the
recipient as detailed on this deed and hand delivered or sent by prepaid post or facsimile to
the number as specified to the sender by notice.
1.9Entire Agreement
This deed embodies the entire agreement between the parties with respect to the subject
matter of this deed and supersedes any prior negotiation, arrangement, understanding or
agreement with respect to the subject matter of any term of this deed.
Page | 15
Execution Page |
EXECUTED AS A DEED
Executed by Telix Pharmaceuticals Limited ACN 616 620 369 in accordance with the Corporations Act 2001 (Cth) by being signed by the following officers: | |||
Director Christian Behrenbruch | Director/Secretary Genevieve Ryan | ||
\ | |||
Name (please print) | Name (please print) |
Signed, sealed and delivered by [insert full name of Director] in the presence of: | |||
[insert full name of Director] | Witness | ||
Name (please print) |

Exhibit 4.8
CERTAIN INFORMATION (INDICATED BY “[***]”) CONTAINED IN THIS AGREEMENT HAS BEEN REDACTED BECAUSE (I) THE
COMPANY CUSTOMARILY AND ACTUALLY TREATS THE REDACTED INFORMATION AS PRIVATE OR CONFIDENTIAL AND (II)
THE OMITTED INFORMATION IS NOT MATERIAL.
Service Agreement
Telix Pharmaceuticals Limited
ACN 616 620 369
and
Dr Christian Peter Behrenbruch





SERVICE AGREEMENT

1Key Terms
SubjectKey Terms
1.PartiesABN 616 620 369 of Suite 226, 55 Flemington Road, North Melbourne,
VIC 3051 (Company)
Dr Christian Peter Behrenbruch of [***] (You)
2.PurposeThe Company has agreed to employ You in the Position and You have agreed to
accept that employment on the terms of this agreement.
3.Position
(see clause 2)
Chief Executive Officer / Executive Director
4.Start Date
(see clause 2.2)
16 January 2017
5.Location
(see clause 2.4)
Melbourne, Australia
6.Manager
(see clause 3.3)
Reporting to the Board of Directors of the Company
7.Total Fixed Remuneration
(see clause 6.1)
$241,000 per year which includes:
(a)base salary; and
(b)compulsory superannuation contributions paid by the Company for Your benefit,
but does not include any payroll tax or workers’ compensation insurance paid by the
Company in respect of Your employment.
8.Review Date
(see clause 6.4)
Remuneration package to be reviewed annually within 30 days of the anniversary of the Start
Date. If the Company executes an initial public offering (IPO) within 12 months of the Start
Date, then the total remuneration package will be reviewed and benchmarked on the basis of
comparable market cap ASX-listed companies.
9.First Review Date
(see clause 6.4)
Within 30 days of 1 January 2018 if the Company remains an unlisted company.
10.Payment Frequency
(see clause 6.5)
Monthly
11.Employee Notice
Period (see clause 9.1(a))
3 months’ notice
1


SubjectKey Terms
12. Employer Notice Period
(see clause 9.1(b))
13.Post Employment
Restraints (see clause 14)
3 months’ notice
(a)No competing with the Group
You must not (whether directly or indirectly) during the Restricted Period (explained
below) and in the Restricted Area (also explained below) carry on, be employed by or
engaged or otherwise interested in any Competitive Business (being any business that
competes with the Group during the period of 6 months preceding the End Date, or
during the Restricted Period) to provide products or services which are the same as or
similar to those You provided to the Company at any time within the 6 months
immediately preceding the date Your employment ends.
(b)No inducing employees to leave the Group
You must not during the Restricted Period and in the Restricted Area induce or attempt
to induce any director or employee of the Group with whom You had work related
dealings during the 6 months preceding the date Your employment ends to terminate his
or her engagement with the Group, whether or not that person would commit a breach of
that person’s contract of engagement.
(c)No persuading the Group’s customer to cease or reduce business
You must not during the Restricted Period and in the Restricted Area solicit or persuade
any customer of the Group with whom You had work related dealings during the 6
months preceding the date Your employment ends to cease doing business with the
Group or reduce the amount of business which the person would normally do, or
otherwise have done, with the Group.
14.Restricted Period
(see clause 14)
(a)Australia, United Kingdom, European Union or United States; or if a Court finds that
unenforceable;
(b)Victoria, Australia; or if a Court finds that unenforceable;
(c)Melbourne, Victoria, Australia.
15.Restricted Period
(see clause 14)
(d)the period 6 months starting on the End Date; or if a Court finds that unenforceable
(e)the period of 3 months starting on the End Date,
where the ‘End Date’ is the date Your employment ends.
16.Governing law and
jurisdiction (see clause 16.1)
Victoria, Australia
2
2Employment and appointment as a director

2.1Position
(a)The Company will employ You in the Position set out in clause 1 (‘Key Details’) at the start of this agreement.
(b)With Your consent, You may also be appointed to the board of directors of the Company or one of its related bodies
corporate. Your appointment will be subject to the relevant company’s constitution.
2.2Start date
Your employment will start on the Start Date set out in clause 1 (‘Key Details’) above.
2.3Warranty
You warrant that You are not under any obligation or restriction which would interfere or conflict with Your employment in this
role or Your obligations and duties under this agreement.
2.4Location and travel
(a)Your place of work is, initially, the Location set out in clause 1 (‘Key Details’) above. The Company may require You to
work in other locations at any time.
(b)The Company may require You to travel within the state, interstate or overseas to perform Your duties.
3Duties and reporting structure

3.1Duties of Your position
(a)You must perform the duties reasonably associated with the Position.
(b)In addition, You must also perform other duties which You are capable of performing, as required by the Company.
3.2General duties
You must:
(a)devote all of Your time, attention and skill to the performance of Your duties both during normal business hours and at
other times as reasonably necessary for a full- time appointment of this nature;
(b)perform Your duties faithfully and diligently;
(c)follow lawful and reasonable directions given to You by the Company; and
(d)promote the interests of the Company and any member of the Group.
3.3Reporting structure
You will report to the Manager set out in clause 1 (‘Key Details’) above or as otherwise directed by the Company.
3.4Changes to Your position
If Your position, duties or reporting structure change, this agreement will continue to apply to Your employment unless You and
the Company:
3
(a)enter a new written employment agreement; or
(b)vary this agreement in writing.
4Hours of work

You are employed for a minimum of 40 hours/week to perform the duties associated with the Position. Because of the senior
nature of the Position, You acknowledge and agree that any hours that You are required to work are reasonable so far as they
are necessary for the full and proper performance of Your duties under this agreement.
5Company policies

The Company has various policies which apply to Your employment. You must familiarise Yourself with these policies. Where
the policies place obligations on You, You must comply with them. The Company may review, vary, add to or withdraw the
policies from time to time in its absolute discretion. To avoid doubt, the policies and any obligations on the Company set out in
them do not form part of Your employment agreement and are not binding on the Company.
6Remuneration
6.1Total fixed remuneration employment cost
(a)The Company will pay You on a Total Fixed Remuneration basis.
(b)Your initial Total Fixed Remuneration is set out in clause 1 (‘Key Details’) above.
(c)The Company will make compulsory superannuation contributions up to the quarterly maximum contribution base on
Your behalf in accordance with Superannuation Guarantee legislation. The contributions may be made to the
Company’s default fund or to a complying superannuation fund of Your choice. You will be given information about this
choice.
(d)You may elect to receive part of Your Total Fixed Remuneration in the form of benefits. The Company’s salary
packaging guidelines set out details of the benefits currently available and the process for electing to receive them. The
Company may withdraw salary packaging at any time in its absolute discretion.
(e)If You elect to receive part of Your Total Fixed Remuneration as benefits, the election must comply with the salary
packaging guidelines. Your Total Fixed Remuneration includes all costs associated with the election, including the
costs of any fringe benefits tax payable by the Company as a result of complying with the election.
6.2Directors and officers insurance
Subject to the applicable law, if You are appointed as a director of the Company or any member of the Group, the Company or
the relevant Group company will maintain directors and officers liability insurance for Your benefit on terms acceptable to the
Company or the relevant Group company.
6.3Annualised salary
Your base salary includes compensation for all entitlements, benefits or payments that might otherwise be due under any
industrial instrument that may apply to Your employment including overtime, penalty payments for out of hours work and
working weekends and public holidays, and any other loadings, penalties, overtime or allowances. Accordingly, You will not be
paid any special rates or allowances for working particular times or under particular conditions unless otherwise agreed in
writing.
4
6.4Review of remuneration package
The Company will review Your Total Fixed Remuneration each year on the Review Date set out in clause 1 (‘Key Details’) above,
with the first review on the First Review Date set out in clause 1 (‘Key Details’) above, unless otherwise agreed in writing.
6.5Payment
The Company will pay any Total Fixed Remuneration that You elect to receive as salary (less any compulsory superannuation
contributions and applicable tax), based on the Payment Frequency set out in clause 1 (‘Key Details’) above, by electronic
transfer to Your nominated bank account.
6.6Expenses
The Company will reimburse You for any expenses that You reasonably incur during the performance of Your duties. The
Company may require You to provide a tax invoice, or other evidence, to substantiate any expenses claim.
7Incentive arrangements

(a)You may be eligible to participate in incentive arrangements offered by the Company from time to time.
(b)Your participation is subject to the rules of the plan. The Company may amend these rules from time to time. They do
not form part of Your employment agreement.
(c)Your participation in the plan, and all benefits under the plan, are at the absolute discretion of the Company. Any
payment to You will be net of any applicable tax and superannuation contribution which the Company is required to
make in respect of any incentive payment.
(d)Unless otherwise agreed in writing, You are only entitled to receive a benefit under the plan if You are employed by the
Company on the date that the benefit is paid or provided.
8Leave

8.1Your entitlement
(a)You are entitled to annual leave, personal/carer’s leave, compassionate leave, and parental leave in accordance with
legislation.
(b)Currently, Your entitlements under the National Employment Standards are 4 weeks’ annual leave each year, 10 days’
paid personal/carer’s leave each year (in certain circumstances You may also be able to access unpaid carer’s leave
and compassionate leave), and up to 12 months’ unpaid parental leave where You have responsibility for the care of a
child (in certain circumstances You may be able to extend this leave).
8.2Long service leave
You will be entitled to long service leave in accordance with applicable legislation.
8.3Public holidays
You will be usually be entitled to paid leave on days declared as public holidays in the state or territory in which You work. From
time to time You may be required, and You will not reasonably refuse, to work on a public holiday in order to perform Your
duties. Your remuneration includes an amount to compensate for this.
5
9Ending Your employment
9.1Ending Your employment with notice
Subject to the notice provisions set out in clause 9.1(a):
(a)You may end Your employment at any time by giving the Employee Notice Period set out in clause 1 (‘Key Details’); or
(b)the Company may end Your employment at any time by giving the Employer Notice Period set out in clause 1 (‘Key
Details’).
9.2Payment in lieu of notice period
The Company may:
(a)pay You in lieu of Your notice period; or
(b)require You to work for part of Your notice period and pay You in lieu of the balance of the period.
9.3Calculation of payments if Your employment ends
If Your employment ends, the Company will calculate any payments in lieu of notice or accrued leave in accordance with
applicable law.
9.4Deduction of amounts owed
(a)To the extent permitted by law, any outstanding advances or other payments due to the Company by You will be
deducted before payment of any amounts under this clause 9 are made to You.
(b)If the amounts owed by You to the Company at the date Your employment ends exceed amounts payable to You under
this clause 9, You agree to repay such amounts to the Company within 14 days of the date Your employment ends.
9.5Duties during notice period
If You or the Company gives notice ending Your employment, the Company may direct You at any time during the notice period:
(a)not to attend work; or
(b)not to perform all or part of Your duties.
9.6Ending Your employment without notice
The Company may end Your employment at any time without notice if You:
(a)engage in serious or wilful misconduct;
(b)are seriously negligent in the performance of Your duties;
(c)commit a serious or persistent breach of this agreement;
(d)commit an act, whether at work or otherwise, which brings the Company into disrepute; or
(e)are charged with an offence punishable by imprisonment.
9.7Return of property
Before Your employment ends, or as soon as practicable after it ends, You must return all property belonging to the Company.
6
9.8Resignation as director
(a)If Your employment ends You must resign as a director of the Company and any Group company unless the Board
determines otherwise.
(b)If You fail to resign as a director You irrevocably appoint the Company Secretary (or any other person nominated by the
Company) as attorney to sign a resignation as director or other officer on Your behalf and have no entitlement to any
compensation for loss of office.
9.9Redundancy
If Your employment ends for a reason including that Your position is redundant, any payment to You on ending of Your
employment (other than a payment in lieu of Your minimum statutory entitlement to notice of termination) will be inclusive of any
statutory entitlement that You may have to redundancy pay.
9.10Consideration for holding office
You and the Company agree that the benefits to which You are entitled under this agreement in connection with the termination
of Your employment are in part consideration for You agreeing to accept the office of the Position, and any other office or
position that is a ‘managerial or executive office’ (as that expression is defined in the Corporations Act 2001 (Cth)) in any Group
company.
10Disclosure of information
10.1Definitions
In this agreement:
(a)‘Confidential Information’ means any Information which is confidential and not in the public domain (unless in the public
domain because of a breach of confidentiality).
(b)‘Information’ means any information about the Group or its business (including, but not limited to, any idea, concept,
process or know-how) which comes to Your notice in the course of Your employment or is generated by You in the
course of performing Your duties.
10.2Importance of Confidential Information
(a)During Your employment with the Company You will acquire experience, Confidential Information, trade secrets, know-
how and particular skills in the affairs, practices, customer requirements and trade connections of the Group.
(b)Because of the importance to the Group of the knowledge and Confidential Information which You will acquire, the
Company wishes to ensure that You do not take advantage of these matters for Your advantage or others to the
detriment of the Group and its businesses and in violation of its rights.
10.3Your obligations during employment
During Your employment, You must not use or disclose Information unless the use or disclosure is:
(a)required by law;
(b)made as part of the proper performance of Your duties; or
(c)agreed by the Company.
7
10.4Your obligations after Your employment ends
After Your employment ends, and without limiting Your general law obligations, You must not disclose Confidential Information
unless the disclosure is:
(a)required by law; or
(b)agreed in writing by the Company.
10.5Preventing disclosure
You must take all reasonable and necessary precautions to maintain the secrecy and prevent disclosure of Information.
10.6Implied term and survival of obligations
(a)To avoid doubt, this clause is not intended to limit any duty of fidelity owed by You and implied into Your employment
agreement.
(b)Your obligations under this clause continue after Your employment ends.
11Intellectual property
11.1Definition
In this clause ‘Intellectual Property’ means all present and future rights to intellectual property including any inventions and
improvements, trade marks (whether registered or common law trade marks), designs, copyright, any corresponding property
rights under the laws of any jurisdiction and any rights in respect of an invention, discovery, trade secret, secret process, know-
how, concept, idea, information, process, data or formula.
11.2Ownership
(a)The Company owns all Intellectual Property that You create or contribute to during Your employment.
(b)You must do all things necessary to ensure that the Company owns Intellectual Property that You create or contribute
to during Your employment.
11.3Disclosure
You must inform the Company of all Intellectual Property that You create or contribute to during Your employment.
11.4Survival of obligations
Your obligations under this clause continue after Your employment ends.
12Moral Rights

(a)In this clause ‘Moral Rights’ means the right of attribution of authorship, the right not to have authorship falsely
attributed and the right of integrity of authorship, as defined in the Copyright Act 1968 (Cth).
(b)If You have Moral Rights in any Intellectual Property owned by the Company, You irrevocably consent to any act or
omission by the Company which infringes those Moral Rights. You agree that Your consent extends to acts and
omissions by the Company’s licensees and successors in title, and You agree that Your consent is a genuine consent
given under Part 9 of the Copyright Act 1968 (Cth) and has not been induced by duress or any false or misleading
statement.
8
(c)Your obligations under this clause continue after Your employment ends.
13Restrictions during Your employment
13.1Other business interests
Subject to clause 13.2, during Your employment You must not be engaged, concerned or interested in any other business
without the Company’s prior written consent, with such consent not to be unreasonably withheld if such business is not in
conflict with clause 3.2 or if disclosed to the Company prior to the commencement of Your employment.
13.2Shareholding
Despite any other clause of this agreement, You may hold shares in companies listed on any recognised stock exchange
without the Company’s prior written consent if You hold less than 5% of the issued shares of any class of any one company.
14Restrictions after Your employment ends
14.1Restrictions
The Post Employment Restraints set out in clause 1 (‘Key Details’) apply to You.
14.2Consent
The Post Employment Restraints do not apply in circumstances where You have obtained the Company’s prior written consent.
14.3Restrictions reasonable and independent
You agree that:
(a)You will obtain Confidential Information during Your employment, the disclosure of which could materially harm the
Group;
(b)the Post Employment Restraints are reasonable and necessary for the protection of the Group’s Confidential
Information and goodwill;
(c)You intend the restrictions to operate to the maximum extent;
(d)damages may be inadequate to protect the Group’s interests and the Group is entitled to seek and obtain injunctive
relief, or any other remedy, in any court; and
(e)the restrictions are separate, distinct and several, so that the unenforceability of any restriction does not affect the
enforceability of the other restrictions.
14.4Modification of restrictions
If the Post Employment Restraints:
(a)are void as unreasonable for the protection of the Group’s interests; and
(b)would be valid if part of the wording was deleted or the period or area was
reduced, the restrictions will apply with the modifications necessary to make them effective.
14.5Obligations continue
Your obligations under the Post Employment Restraints and this clause 14 survive the ending of Your employment.
9
15Compliance and approvals
(a)The exercise of, or compliance with, any discretion, right or obligation under this agreement is subject to any required
board or shareholder approvals, any necessary regulatory consent and compliance with the Company’s constitution
and all applicable laws.
(b)Notwithstanding any provision of this agreement, the Company is not required to pay or provide, or procure the payment
or provision, of any payment or benefit to You which is not permitted by the provisions of Part 2D.2, Division 2 or
Chapter 2E of the Corporations Act 2001 (Cth) in the absence of shareholder approval. Any such payments or benefits
must be reduced to ensure compliance with this clause and there is no obligation on the Company to seek or obtain
shareholder approval. In the event of overpayment, You must, on receiving written notice from the Company Secretary
(or his or her nominee), immediately repay any monies or benefits specified in such notice.
(c)This clause 15 has effect regardless of any other provision of this agreement.
16General

16.1Governing law and jurisdiction
(a)This agreement is governed by the law in force in Governing law and jurisdiction in clause 1 (‘Key Details’) above.
(b)Each party irrevocably submits to the non-exclusive jurisdiction of courts of the jurisdiction specified in clause 1 (‘Key
Details’) above.
16.2Entire agreement and no reliance
(a)This agreement states all the express terms of the agreement between the parties in respect of its subject matter. It
supersedes all prior discussions, negotiations, understandings and agreements in respect of its subject matter.
(b)You acknowledge that in accepting employment with the Company You have not relied on any representations
regarding Your employment made by the Company (or its agents or employees) other than matters expressly set out in
this agreement.
16.3Legal advice
You represent that You have taken, or had the opportunity of taking, legal advice in relation to the nature, effect and extent of
this agreement.
16.4Counterparts
This agreement may be executed in any number of counterparts and all counterparts, taken together, constitute one instrument.
A party may execute this agreement by executing any counterpart.
16.5Benefit of this agreement
The Company executes this agreement for the Group. You acknowledge and agree that each Group Company may
independently enforce the obligations given in their favour in this agreement against You in their own right.
16.6Interpretation
In this agreement:
(a)A reference to the ‘Group’ means the Company and each ‘related body corporate as that expression is defined in the
Corporations Act 2001 (Cth).
10
(b)Headings and bold type are for convenience only and do not affect the interpretation of this agreement.
(c)The singular includes the plural and the plural includes the singular.
(d)Words of any gender include all genders.
(e)Other parts of speech and grammatical forms of a word or phrase defined in this agreement have a corresponding
meaning.
(f)An expression importing a person includes any company, partnership, joint venture, association, corporation or other
body corporate and any government agency as well as an individual.
(g)A reference to a clause, party, schedule, attachment or exhibit is a reference to a clause of, and a party, schedule,
attachment or exhibit to, this agreement.
(h)A reference to any legislation includes all delegated legislation made under it and amendments, consolidations,
replacements or re-enactments of any of them.
(i)No provision of this agreement will be construed adversely to a party because that party was responsible for the
preparation of this agreement or that provision.
(j)Specifying anything in this agreement after the words ‘include’ or ‘for example’ or similar expressions does not limit
what else is included.
(k)This agreement includes any schedule.
11
Signing Page
Executed as an agreement
COMPANY
EXECUTED on behalf of:
TELIX PHARMACEUTICALS LIMITED by:
/s/ Michael Cawley
Signature of Director

Signature of Company Secretary/Director
Michael Cawley
Director (please print)

Company Secretary/Director (please print)
YOU
EXECUTED by DR CHRISTIAN PETER BEHRENBRUCH in the
presence of:
/s/ Evgeniya Mikulchik
Signature of witness
Evgeniya Mikulchik
Name of witness (please print)
/s/ Christian Behrenbruch
Signature of Dr Christian Peter Behrenbruch

Exhibit 4.9
CERTAIN INFORMATION (INDICATED BY “[***]”) CONTAINED IN THIS AGREEMENT HAS BEEN REDACTED BECAUSE (I) THE
COMPANY CUSTOMARILY AND ACTUALLY TREATS THE REDACTED INFORMATION AS PRIVATE OR CONFIDENTIAL AND (II)
THE OMITTED INFORMATION IS NOT MATERIAL.

Employment Agreement
Telix Pharmaceuticals limited
ACN 616 620 369
and
Darren Smith
EMPLOYMENT AGREEMENT


1KEY DETAILS
1.1Parties
Telix Pharmaceuticals Limited ABN 616 620 369 of Suite 401, 55 Flemington Road, North Melbourne, VIC 3051(Company)
Darren Smith of the address set out in the Offer of Employment (You).
Employment
1.2Position
The Company will employ You in the Position set out in the Offer of Employment.
1.3Start date
Your employment will start on the Start Date set out in the Offer of Employment.
You will be subject to the Probation Period set out in the Offer of Employment.
1.4Warranty
You warrant that You are not under any obligation or restriction which would interfere or conflict with Your employment in this role or Your
obligations and duties under this agreement. Your employment with the Company is at all times conditional upon:
(a)You obtaining and retaining all necessary visas, work permits, licences, registrations, or memberships to enable You to lawfully
reside and work in Australia and fulfil the duties of the Position; and
(b)You being competent to properly carry out the duties of the Position and that any representations as to the qualifications, skills,
experience, industry knowledge, business influence, client contacts, and employment history made by You or a person on Your
behalf are true and correct.
1.5Location and travel
(a)Your place of work is, initially, the Location set out in the Offer of Employment. The Company may require You to work in other
locations at any time.
(b)The Company may require You to travel within the state, interstate or overseas to perform Your duties without any additional
remuneration.
2DUTIES AND REPORTING STRUCTURE
2.1Duties of Your position
(a)You must perform the duties reasonably associated with the Position.
(b)In addition, You must also perform other duties which You are capable of performing, as required and directed by the Company.
(c)Your role will include, but not be limited to, the Key Activities set out in the position description.
2.2General duties
You must:
(a)devote Your time, attention and skill to the performance of Your duties as is reasonably necessary for an appointment of this
nature;
(b)perform Your duties faithfully and diligently;
(c)follow lawful and reasonable directions given to You by the Company; and
(d)promote the interests of the Company and any member of the Group.
2.3Reporting structure
You will report to the Manager set out in the Offer of Employment or as otherwise directed by the Company.
2.4Changes to Your position
If Your position, duties or reporting structure change, this agreement will continue to apply to Your employment unless You and the
Company:
(a)enter a new written employment agreement; or
(b)vary this agreement in writing.
2.5Continuous development
You agree to participate in:
(a)Development and/or training programs from time to time determined by the CEO or the CEO’s delegate, at the expense of the
Company;
(b)processes of internal and external review and benchmarking of performance as may be determined to be appropriate by the CEO
or the CEO’s delegate; and
(c)training and readiness in working in a public listed company.
3HOURS OF WORK
Your hours of work are as set out in the Offer of Employment.
From time to time you may be required to work outside your normal hours of work in order to meet the organisation’s international
business needs and your individual duties and objectives. This has been taken into account in calculating your remuneration and you will
not receive additional remuneration for that work.
4COMPANY POLICIES
4.1General
The Company has various policies (including a Code of Conduct) which apply to Your employment. You must familiarise Yourself with
these policies. Where the policies place obligations on You, they constitute a lawful and reasonable direction from the Company and You
must comply with them. The Company may review, vary, add to, apply, not apply or withdraw the policies from time to time in its absolute
discretion. To avoid doubt, the policies and any obligations on the Company set out in them do not form part of Your employment
agreement and are not binding on the Company.
4.2Workplace Health and Safety
You must attend to Your work safety and notify Your Manager if you become aware of any workplace risks. You must comply with all work
health and safety policies of the Company.
4.3Anti-Discrimination, Bullying and Harassment
The Company is an equal opportunity employer. You must comply at all times with the Company’s policies in respect of anti-
discrimination, bullying and harassment.
5REMUNERATION
5.1Total remuneration employment cost
(a)Your initial Total Remuneration is set out in the Offer of Employment..
(b)The Company will make compulsory superannuation contributions up to the quarterly maximum contribution base on Your behalf
in accordance with Superannuation Guarantee legislation. The contributions may be made to the Company’s default fund or to a
complying superannuation fund of Your choice. You will be given information about this choice. If You do not nominate a
complying superannuation fund for this purpose, the contribution may be paid into an existing stapled account identified by the
Commissioner of Taxation.
5.2Annualised salary
Your base salary absorbs all entitlements, benefits and payments that You may now have or subsequently acquire. Your base salary
includes compensation for all entitlements, benefits or payments that might otherwise be due under the Fair Work Act 2009 (Cth) (FW
Act) or any industrial instrument that may apply to Your employment including overtime and penalty payments for out of hours work and
working weekends and public holidays, and any other loadings, penalties, overtime or allowances. Accordingly, You will not be paid any
special rates or allowances for working particular times or under particular conditions unless otherwise agreed in writing. Further, any
remuneration paid in excess of any particular entitlement may be offset against any other entitlement, including an entitlement in a different
pay period.
5.3Guarantee of annual earnings
The Company undertakes that the base salary and any other benefits paid to you is a guarantee of annual earnings for the purposes of the
FW Act for a period of at least 12 months. You accept that undertaking and acknowledge that Your employment will not be subject to the
application of any modern award for any period during which you earn in excess of the high income threshold as defined by the FW Act.
5.4Payment
The Company will pay any Total Remuneration that You elect to receive as salary (less any compulsory superannuation contributions and
applicable tax), based on the Payment Frequency set out in the Offer of Employment, by electronic transfer to Your nominated bank
account.
5.5Expenses
The Company will reimburse You for any expenses that You reasonably incur during the performance of Your duties. The Company may
require You to provide a tax invoice, or other evidence, to substantiate any expenses claim.
6INCENTIVE ARRANGEMENTS
Long Term Incentives
(a)You may be eligible to participate in long-term incentive arrangements offered by the Company from time to time.
(b)Your participation is subject to the rules of the plan. The Company may amend or discontinue the plan and the rules of the plan
from time to time. They do not form part of Your employment agreement.
(c)Any payment made to You under the plan will not give rise to an expectation or create any precedent for the awarding of any
subsequent payment(s).
(d)Your participation in the plan, and all benefits under the plan, are at the absolute discretion of the Company. Any payment to You
will be net of any applicable tax and superannuation contribution which the Company is required to make in respect of any
incentive payment.
(e)Unless otherwise agreed in writing, You are only entitled to receive a benefit under the plan if You are employed by the Company
and not subject to a notice of termination period on the date that the benefit is paid or provided.
Short Term Incentives
(a)You may be eligible to participate in any short-term incentive plans of the Board, which may be determined by the Board from time
to time.
(b)You may be entitled to a bonus incentive of up to a portion of your Base Salary set out in the Offer of Employment, which, if
payable, will be payable based on Your performance against agreed performance milestones and may be paid annually in cash or
equity and at the Board’s discretion.
(c)Your eligibility and any payment is subject to the rules of any plan or policy in place from time to time. The Board may amend or
discontinue the plan and the rules of the plan from time to time. They do not form part of Your employment agreement.
(d)Any payment made to You under the plan will not give rise to an expectation or create any precedent for the awarding of any
subsequent payment(s).
(e)Your participation in the plan, and all benefits under the plan, are at the absolute discretion of the Board. Any payment to You will
be net of any applicable tax and superannuation contribution which the Company is required to make in respect of any incentive
payment.
(f)Unless otherwise agreed in writing, You are only entitled to receive a benefit under the plan if You are employed by the Company
and not subject to a notice of termination period on the date that the benefit is paid or provided.
7LEAVE
7.1Your entitlement
(a)You are entitled to annual leave, personal/carer’s leave, compassionate leave, and parental leave in accordance with legislation.
(b)Currently, Your entitlements under the National Employment Standards are 4 weeks annual leave each year, 10 days paid personal/
carer’s leave each year (in certain circumstances You may also be able to access unpaid carer’s leave), compassionate leave,
and up to 12 months unpaid parental leave where You have responsibility for the care of a child (in certain circumstances You may
be able to extend this leave).
(c)Entitlements for part-time employees are pro-rated accordingly.
You must notify your Manager immediately before taking personal/carer’s leave so that the Company may accommodate your absence. If
this is not possible, You must contact your Manager or the Company as soon as you are able to. The Company may require you to
provide a medical certificate or attend a medical examination at any time.
7.2Long service leave
You will be entitled to long service leave in accordance with applicable legislation.
7.3Public holidays
You will usually be entitled to paid leave on days declared as public holidays in the state or territory in which You work. From time to time
You may be required, and You will not reasonably refuse, to work on a public holiday in order to perform Your duties. Your remuneration
includes an amount to compensate for this.
8ENDING YOUR EMPLOYMENT
8.1Ending Your employment with notice
After the Probation Period and subject to clause 8.6:
(a)You may end Your employment at any time by giving the Employee Notice Period set out in the Offer of Employment; or
(b)the Company may end Your employment at any time by giving the Employer Notice Period set out in the Offer of Employment.
8.2Payment in lieu of notice period
In the event that either You or the Company provide notice in accordance with clause 8.1 above, the Company may elect to:
(a)pay You in lieu of Your notice period; or
(b)require You to work for part of Your notice period and pay You in lieu of the balance of the period.
8.3Calculation of payments if Your employment ends
If Your employment ends, the Company will calculate any payments in lieu of notice or accrued leave in accordance with applicable law.
8.4Deduction of amounts owed
(a)To the extent permitted by law, any outstanding advances or other payments due to the Company by You will be deducted before
payment of any amounts under this clause 8 are made to You.
(b)If the amounts owed by You to the Company at the date Your employment ends exceed amounts payable to You under this clause
8, You agree to repay such amounts to the Company within 14 days of the date Your employment ends.
8.5Duties during notice period
If You or the Company gives notice ending Your employment, the Company may direct You at any time during the notice period:
(a)not to attend work;
(b)not to perform all or part of Your duties;
(c)to perform duties which are different to Your usual duties, provided that You have the necessary skills and competencies to
perform these duties;
(d)to assist the Company with a proper hand over of the duties of the Position including business information, work, clients, and
business;
(e)not to have any dealings with any customers, suppliers or clients of the Company or the Group; or
(f)to do any combination of the above.
8.6Ending Your employment without notice
The Company may end Your employment at any time without notice if You:
(a)engage in serious or wilful misconduct;
(b)are seriously negligent in the performance of Your duties;
(c)commit a serious or persistent breach of Company policy (including but not limited to a serious or persistent beach of the Code of
Conduct);
(d)commit a serious or persistent breach of this agreement;
(e)refuse to carry out lawful and reasonable instructions of the Company;
(f)commit an act, whether at work or otherwise, which brings the Company into disrepute; or
(g)are charged with an offence punishable by imprisonment.
8.7Suspension
If the Company suspects that You have been involved in any improper conduct or involved in any other conduct which in the opinion of
the Company may impact upon Your ability to carry out Your duties and responsibilities under this agreement or may cause damage to the
Company’s business or reputation, the Company may do any or all of the following:
(a)suspend You from performing the duties and responsibilities of the Position for a period determined by the Company;
(b)direct You not to attend the workplace, communicate with fellow employees, customers, suppliers or clients of the Company or
any other persons involved in the conduct which is being investigated, or otherwise interfere with the conduct of the
investigation; and
(c)appoint any person to conduct the investigation; and direct You to provide any assistance and answer any questions required
for the investigation.
During the period of suspension You will continue to receive Your Total Remuneration under this agreement. Any suspension under this
clause will not be treated as disciplinary action by the Company, but will be instituted solely for the purpose of conducting an
investigation.
8.8Return of property
Before Your employment ends, or as soon as practicable after it ends, You must return all property belonging to the Company.
8.9Redundancy
If Your employment ends for a reason including that Your position is redundant, any payment to You on ending of Your employment (other
than a payment in lieu of Your minimum statutory entitlement to notice of termination) will be inclusive of any statutory entitlement that
You may have to redundancy pay.
8.10Consideration for holding office
You and the Company agree that the benefits to which You are entitled under this agreement in connection with the termination of Your
employment are in part consideration for You agreeing to accept the office of the Position, and any other office or position that is a
‘managerial or executive office’ (as that expression is defined in the Corporations Act 2001 (Cth)) in any Group company.
9DISCLOSURE OF INFORMATION
9.1Definitions
In this agreement:
(a)‘Confidential Information’ means any Information which is confidential and not in the public domain (unless in the public domain
because of a breach of confidentiality) including, but not limited to:
(i)the business or affairs, financial information, Intellectual Property, and sales and marketing information of the Company
or Group, or their respective customers or suppliers;
(ii)information which is marked “confidential” or which is described or treated by the Company or Group as confidential;
(iii)information of a business sensitive nature; and
(iv)trade secrets, research and confidential information and know-how of the Company, the Group, or their respective
customers or suppliers.
(b)‘Information’ means any information about the Group or its business (including, but not limited to, any idea, concept, process or
know-how) which comes to Your notice in the course of Your employment or is generated by You in the course of performing
Your duties.
9.2Importance of Confidential Information
(a)During Your employment with the Company You will acquire experience, Confidential Information, trade secrets, know-how and
particular skills in the affairs, practices, customer requirements and trade connections of the Group.
(b)Because of the importance to the Group of the knowledge and Confidential Information which You will acquire, the Company
wishes to ensure that You do not take advantage of these matters for Your advantage or others to the detriment of the Group and
its businesses and in violation of its rights.
9.3Your obligations during employment
During Your employment, You must not use or disclose Information unless the use or disclosure is:
(a)required by law;
(b)made as part of the proper performance of Your duties; or
(c)agreed in writing by the Company.
9.4Your obligations after Your employment ends
After Your employment ends, and without limiting Your general law obligations, You must not disclose Confidential Information unless the
disclosure is:
(a)required by law; or
(b)agreed in writing by the Company.
9.5Preventing disclosure
You must take all reasonable and necessary precautions to maintain the secrecy and prevent disclosure of Information. You must
immediately notify the Company if You suspect or become aware that Confidential Information has been improperly copied, used or
disclosed.
9.6Implied term and survival of obligations
(a)To avoid doubt, this clause is not intended to limit any duty of fidelity owed by You and implied into Your employment
agreement.
(b)Your obligations under this clause continue after Your employment ends.
10INTELLECTUAL PROPERTY
10.1Definition
In this clause ‘Intellectual Property’ means all present and future rights to intellectual property including any inventions and
improvements, trade marks (whether registered or common law trademarks), designs, copyright, patents, any corresponding property
rights under the laws of any jurisdiction and any rights in respect of any work, including any invention, discovery, trade secret, secret
process, know-how, concept, idea, information, process, data or formula.
10.2Ownership
(a)The Company owns all Intellectual Property that You create or contribute to during Your employment.
(b)You must do all things necessary to ensure that the Company owns Intellectual Property that You create or contribute to during
Your employment.
10.3Disclosure
You must inform the Company of all Intellectual Property that You create or contribute to during Your employment.
10.4Survival of obligations
Your obligations under this clause continue after Your employment ends.
11MORAL RIGHTS
(a)In this clause ‘Moral Rights’ means the right of attribution of authorship, the right not to have authorship falsely attributed and
the right of integrity of authorship, as defined in the Copyright Act 1968 (Cth).
(b)If You have Moral Rights in any Intellectual Property owned by the Company, You irrevocably consent to any act or omission by
the Company which infringes those Moral Rights. You agree that Your consent extends to acts and omissions by the Company’s
licensees and successors in title, and You agree that Your consent is a genuine consent given under Part 9 of the Copyright Act
1968 (Cth) and has not been induced by duress or any false or misleading statement.
(c)Your obligations under this clause continue after Your employment ends.
12RESTRICTIONS DURING YOUR EMPLOYMENT
12.1Other business interests
Subject to clause 12.2, during Your employment You must not be engaged, concerned or interested in any other business without the
Company’s prior written consent, with such consent not to be unreasonably withheld if such business is not in conflict with clause 2.2 or
if disclosed to the Company prior to the commencement of Your employment.
12.2Shareholding
Despite any other clause of this agreement, You may hold shares in companies listed on any recognised stock exchange without the
Company’s prior written consent if You hold less than 5% of the issued shares of any class of any one company.
13RESTRICTIONS AFTER YOUR EMPLOYMENT ENDS
13.1Restrictions
The Post Employment Restraints set out in the Offer of Employment apply to You.
13.2Consent
The Post Employment Restraints do not apply in circumstances where You have obtained the Company’s prior written consent.
13.3Restrictions reasonable and independent
You agree that:
(a)You will obtain Confidential Information during Your employment, the disclosure of which could materially harm the Group;
(b)You will develop influence over customers, directors, employees and/or contractors of the Group;
(c)the Post Employment Restraints are reasonable and necessary for the protection of the Group’s Confidential Information,
goodwill and relationships with its customers, directors, employees and contractors;
(d)You intend the restrictions to operate to the maximum extent;
(e)damages may be inadequate to protect the Group’s interests and the Group is entitled to seek and obtain injunctive relief, or any
other remedy, in any court; and
(f)the restrictions are separate, distinct and several, so that the unenforceability of any restriction does not affect the enforceability
of the other restrictions.
13.4Modification of restrictions
If the Post Employment Restraints:
(a)are void as unreasonable for the protection of the Group’s interests; and
(b)would be valid if part of the wording was deleted or the period or area was
reduced, the restrictions will apply with the modifications necessary to make them effective.
13.5Obligations continue
Your obligations under the Post Employment Restraints and this clause 13 survive the ending of Your employment.
14COMPLIANCE AND APPROVALS
(a)The exercise of, or compliance with, any discretion, right or obligation under this agreement is subject to any required board or
shareholder approvals, any necessary regulatory consent and compliance with the Company’s constitution and all applicable
laws.
(b)Notwithstanding any provision of this agreement, the Company is not required to pay or provide, or procure the payment or
provision, of any payment or benefit to You which is not permitted by the provisions of Part 2D.2, Division 2 or Chapter 2E of the
Corporations Act 2001 (Cth) in the absence of shareholder approval. Any such payments or benefits must be reduced to ensure
compliance with this clause and there is no obligation on the Company to seek or obtain shareholder approval. In the event of
overpayment, You must, on receiving written notice from the Company Secretary (or his or her nominee), immediately repay any
monies or benefits specified in such notice.
(c)This clause 14 has effect regardless of any other provision of this agreement.
15GENERAL
15.1Governing law and jurisdiction
(a)This agreement is governed by the law in force in Governing law and jurisdiction in the Offer of Employment .
(b)Each party irrevocably submits to the non-exclusive jurisdiction of courts of the jurisdiction specified in the Offer of Employment
.
15.2Entire agreement and no reliance
(a)This agreement states all the express terms of the agreement between the parties in respect of its subject matter. It supersedes all
prior discussions, negotiations, understandings and agreements in respect of its subject matter.
(b)You acknowledge that in accepting employment with the Company You have not relied on any representations regarding Your
employment made by the Company (or its agents or employees) other than matters expressly set out in this agreement.
(c)This agreement may only be amended by agreement in writing signed by both parties.
15.3Legal advice
You represent that You have taken, or had the opportunity of taking, legal advice in relation to the nature, effect and extent of this
agreement.
15.4Counterparts
This agreement may be executed in any number of counterparts and all counterparts, taken together, constitute one instrument. A party
may execute this agreement by executing any counterpart.
15.5Benefit of this agreement
The Company executes this agreement for the Group. You acknowledge and agree that each Group Company may independently enforce
the obligations given in their favour in this agreement against You in their own right.
15.6Interpretation
In this agreement:
(a)A reference to the ‘Group’ means the Company and each ‘related body corporate’ as that expression is defined in the
Corporations Act 2001 (Cth).
(b)Headings and bold type are for convenience only and do not affect the interpretation of this agreement.
(c)The singular includes the plural and the plural includes the singular.
(d)Words of any gender include all genders.
(e)Other parts of speech and grammatical forms of a word or phrase defined in this agreement have a corresponding meaning.
(f)An expression importing a person includes any company, partnership, joint venture, association, corporation or other body
corporate and any government agency as well as an individual.
(g)A reference to a clause, party, schedule, attachment or exhibit is a reference to a clause of, and a party, schedule, attachment or
exhibit to, this agreement.
(h)A reference to any legislation includes all delegated legislation made under it and amendments, consolidations, replacements or
re-enactments of any of them.
(i)No provision of this agreement will be construed adversely to a party because that party was responsible for the preparation of
this agreement or that provision.
(j)Specifying anything in this agreement after the words ‘include’ or ‘for example’ or similar expressions does not limit what else is
included.
(k)This agreement includes any schedule.
SIGNING PAGE
Executed as an agreement
COMPANY
EXECUTED on behalf of:
TELIX PHARMACEUTICALS LIMITED by:
/s/ Doug CubbinDoug Cubbin
Signature of authorised representativeName of authorised representative
/s/ David CadeDavid Cade
Signature of Regional PresidentName of Regional President
/s/ Helen HovengaHelen Hovenga
Signature of witnessName of witness
YOU
EXECUTED by Darren Smith in the presence of:
/s/ Sarah Louise McRae/s/ Darren Smith
Signature of witnessSignature of Darren Smith
Sarah Louise McRae
Name of witness (please print)
Telix Pharmaceuticals Ltd
Suite 401, 55 Flemington Road
North Melbourne
Victoria, 3051
Australia

18 Jan, 2022
Private and confidential
Darren Smith
[***]
Offer of Employment
Dear Darren,
We are pleased to offer you employment with Telix Pharmaceuticals, as set out below. Terms and conditions of this appointment are conditional of
acceptance of the attached Employment Agreement.
Key Details | ||
1. | Position Title | Deputy Group Chief Financial Officer |
2. | Employment Type | Full time |
3. | Contract Type | Permanent |
4. | Employment Dates | Start Date: Jan 31, 2022 Probation Period End date: Jun 30, 2022 |
5. | Contracted Hours | 37.5 hours per week (1 FTE) This is based on working five normal working days of 7.5 hours each day. |
6. | Location | Primarily based in New South Whales with travel as required to the Company’s head office, currently located at Suite 401, 55 Flemington Road, North Melbourne, Victoria, Australia. |
7. | Key Activities | Refer to attached Position Description |
8. | Manager | Group Chief Financial Officer |
9. | Total Remuneration | (a)base salary of $250,000 AUD per annum; plus (b)compulsory superannuation contributions paid by the Company for Your benefit, but does not include any payroll tax or workers’ compensation insurance paid by the Company in respect of Your employment. |

Telix Pharmaceuticals Ltd | ACN 616 620 369
Key Details | ||
10. | Payment Frequency | Monthly |
11. | Short Term Incentive Rate | You may be eligible to receive a short term incentive of up to 20% of your base salary, subject to the terms set out in the Employment Agreement. Whether the payment is made and the amount of any payment is in the absolute discretion of the Board and the Company. |
12. | Employee Notice Period | 3 months’ notice |
13. | Employer Notice Period | 3 months’ notice |
14. | Post Employment Restraints | (a)No competing with the Group Unless the Company provides prior written consent, You must not (whether directly or indirectly) during the Restricted Period (explained below) and in the Restricted Area (also explained below) carry on, be employed by or engaged or otherwise interested in any Competitive Business (being any business that competes with the Group during the period of 3 months preceding the End Date, or during the Restricted Period) to provide products or services which are the same as or similar to those You provided to the Company at any time within the 3 months immediately preceding the date on which Your employment ends. (b)No inducing employees or contractors to leave the Group You must not during the Restricted Period and in the Restricted Area induce or attempt to induce any director, employee or contractor of the Group with whom You had work related dealings during the 3 months preceding the date on which Your employment ends to terminate his or her employment or engagement with the Group, whether or not that person would commit a breach of that person’s contract of employment or engagement. (c)No persuading the Group’s customer to cease or reduce business You must not during the Restricted Period and in the Restricted Area solicit or persuade any customer of the Group with whom You had work related dealings during the 3 months preceding the date on which Your employment ends to cease doing business with the Group or reduce the amount of business which the person would normally do, or otherwise have done, with the Group. |
15. | Restricted Area | (a)Australia; or if a Court finds that unenforceable (b)Victoria, Australia; or if a Court finds that unenforceable (c)Melbourne, Victoria, Australia. |
16. | Restricted Period | (a)the period of 3 months starting on the End Date; or if a Court finds that unenforceable (b)the period of 1 month starting on the End Date, where the ‘End Date’ is the date Your employment ends. |
Page 2
Key Details | ||
17. | Governing law and jurisdiction | Victoria, Australia |
18. | Review Date | Your Total Remuneration package will be reviewed annually as part of the Company’s remuneration review process. In undertaking this review, the Company may have regard to any matter in its absolute discretion. This review will not necessarily lead to an increase in Your Total Remuneration. |
19. | Probation Period | 6 months’ probation, commencing on Start Date. During this time either party can terminate this contract with two weeks’ notice. |
We look forward to having you join us at Telix and contributing to our vision of helping people with cancer and rare disease live longer, better
quality lives.
Executed as an agreement
COMPANY
EXECUTED on behalf of:
TELIX PHARMACEUTICALS LIMITED by:
/s/ Doug CubbinDoug Cubbin
Signature of authorised representativeName of authorised


representative
/s/ David CadeDavid Cade
Signature of Regional PresidentName of Regional President
/s/ Helena HovengaHelena Hovenga
Signature of witnessName of witness
Page 3
YOU
EXECUTED by Darren Smith in the presence of:
/s/ Sarah Louise Mcrae/s/ Darren Smith
Signature of witnessSignature of Darren Smith
SARAH LOUISE MCRAE
Name of witness (please print)
Page 4

July 25, 2022
Private and confidential
Darren Smith
[***]
Promotion to Group Chief Financial Officer
Dear Darren,
I am pleased to confirm your promotion with Telix Pharmaceuticals, as set out below.
Exhibit 10.10.1
Telix Pharmaceuticals Ltd
Suite 401, 55 Flemington Road
North Melbourne
Victoria, 3051
Australia
Position Title | Group Chief Financial Officer |
Employment Type | Full time |
Contract Type | Permanent |
Effective Dates | Start Date: Aug 01, 2022 |
Contracted Hours | 37.5 hours per week (1.0 FTE) This is based on working five normal working days of 7.5 hours each day. |
Manager | Managing Director and Group CEO |
Reports of Position | Direct: POS0216 - Chief Information Officer POS0065 - Chief Governance and Risk Officer POS0041 - Chief People Officer POS0056 - General Counsel POS0025 - Global Director of Finance POS0040 - CFO - APAC POS0037 - CFO - EMEA POS0117 - CFO Americas |
Total Fixed Remuneration | (a)base salary of $400,000 AUD per annum; plus (b)compulsory superannuation contributions paid by the Company for Your benefit, but does not include any payroll tax or workers’ compensation insurance paid by the Company in respect of Your employment. |
Telix Pharmaceuticals Ltd │A0CAN 616 620 369
Short Term Incentive | At the discretion of Telix Pharmaceuticals and subject to all relevant terms from your original employment agreement and STVR letters issued to you, your Short-Term Variable Remuneration may be up to 27% of your base salary. |
Long Term Incentive | At the discretion of Telix Pharmaceuticals and subject to all relevant terms from your original employment agreement and LTVR letters issued to you, your Long-Term Variable Renumeration may be up to 35% of your base salary. |
Employee Notice Period | 4 months’ notice |
Employer Notice Period | 4 months’ notice |
Post-Employment Restricted Area | (a)Australia; or if a Court finds that unenforceable (b)Victoria, Australia; or if a Court finds that unenforceable (c)Melbourne, Victoria, Australia |
Post-Employment Restricted Period | (a)the period of 6 months starting on the End Date; or if a Court finds that unenforceable (b)the period of 3 months starting on the End Date; or if a Court finds that unenforceable (c)the period of 1 month starting on the End Date, where the ‘End Date’ is the date Your employment ends. |
All other terms and conditions will be in accordance with your original employment letter and agreement, unless varied by any subsequent
employment letters.
Page 2
We look forward to your continued contribution to our purpose of helping people with cancer and rare diseases live longer, better quality lives.
EXECUTED on behalf of:
TELIX PHARMACEUTICALS LIMITED by:
/s/ Chris BehrenbruchChris Behrenbruch
Signature of Managing Director and Group CEOManaging Director


and Group CEO
I, Darren Smith, acknowledge that I have read and accept the terms and conditions of my employment as set out above.
YOU
/s/ Darren Smith28/7/2022
Signature
Date
Page 3

Exhibit 4.10
CERTAIN INFORMATION (INDICATED BY “[***]”) CONTAINED IN THIS AGREEMENT HAS BEEN
REDACTED BECAUSE (I) THE COMPANY CUSTOMARILY AND ACTUALLY TREATS THE
REDACTED INFORMATION AS PRIVATE OR CONFIDENTIAL AND (II) THE OMITTED INFORMATION
IS NOT MATERIAL.

Employment Agreement
Telix Pharmaceuticals (Corporate) Pty Ltd
ACN 666 576 343
and
David Cade

KEY DETAILS

1.1Parties
Telix Pharmaceuticals (Corporate) Pty Ltd ABN 84 666 576 343 of Level 4, 55 Flemington Road, North Melbourne,
VIC 3051 (Company)
David Cade of the address set out in the Offer of Employment (You).
Employment
1.2Position
The Company will employ You in the Position set out in the Offer of Employment.
1.3Start date
Your employment will start on the Start Date set out in the Offer of Employment.
You will be subject to the Probation Period set out in the Offer of Employment.
1.4Warranty
You warrant that You are not under any obligation or restriction which would interfere or conflict with Your employment
in this role or Your obligations and duties under this agreement. Your employment with the Company is at all times
conditional upon:
(a)You obtaining and retaining all necessary visas, work permits, licences, registrations, or memberships to enable You to lawfully
reside and work in Australia and fulfil the duties of the Position; and
(b)You being competent to properly carry out the duties of the Position and that any representations as to the qualifications, skills,
experience, industry knowledge, business influence, client contacts, and employment history made by You or a person on Your
behalf are true and correct.
1.5Location and travel
(a)Your place of work is, initially, the Location set out in the Offer of Employment. The Company may require You to work in other
locations at any time.
(b)The Company may require You to travel within the state, interstate or overseas to perform Your duties without any additional
remuneration.
2DUTIES AND REPORTING STRUCTURE
2.1Duties of Your position
(a)You must perform the duties reasonably associated with the Position.
(b)In addition, You must also perform other duties which You are capable of performing, as required and directed by the Company.
(c)Your role will include, but not be limited to, the Key Activities set out in the position description.
2.2
2.3General duties
You must:
(a)devote Your time, attention and skill to the performance of Your duties as is reasonably necessary for an appointment of this
nature;
(b)perform Your duties faithfully and diligently;
(c)follow lawful and reasonable directions given to You by the Company; and
(d)promote the interests of the Company and any member of the Group.
2.4Reporting structure
You will report to the Manager set out in the Offer of Employment or as otherwise directed by the Company.
2.5Changes to Your position
If Your position, duties or reporting structure change, this agreement will continue to apply to Your employment unless
You and the Company:
(a)enter a new written employment agreement; or
(b)vary this agreement in writing.
2.6Continuous development
You agree to participate in:
(a)Development and/or training programs from time to time determined by the CEO or the CEO’s delegate, at the expense of the
Company;
(b)processes of internal and external review and benchmarking of performance as may be determined to be appropriate by the CEO
or the CEO’s delegate; and
(c)training and readiness in working in a public listed company.
3HOURS OF WORK
Your hours of work are as set out in the Offer of Employment.
From time to time you may be required to work outside your normal hours of work in order to meet the organisation’s
international business needs and your individual duties and objectives. This has been taken into account in calculating
your remuneration and you will not receive additional remuneration for that work.
4COMPANY POLICIES
4.1General
The Company has various policies (including a Code of Conduct) which apply to Your employment. You must familiarise
Yourself with these policies. Where the policies place obligations on You, they constitute a lawful and reasonable
direction from the Company and You must comply with them. The Company may review, vary, add to, apply, not apply
or withdraw the policies from time to time in its absolute discretion. To avoid doubt, the policies and any obligations on
the Company set out in them do not form part of Your employment agreement and are not binding on the Company.
4.2Workplace Health and Safety
You must attend to Your work safety and notify Your Manager if you become aware of any workplace risks. You must
comply with all work health and safety policies of the Company.
4.3Anti-Discrimination, Bullying and Harassment
The Company is an equal opportunity employer. You must comply at all times with the Company's policies
in respect of anti-discrimination, bullying and harassment.
5REMUNERATION
5.1Total remuneration employment cost
(a)Your initial Total Remuneration is set out in the Offer of Employment.
(b)The Company will make compulsory superannuation contributions up to the quarterly maximum contribution base on Your
behalf in accordance with Superannuation Guarantee legislation. The contributions may be made to the Company’s default fund
or to a complying superannuation fund of Your choice. You will be given information about this choice. If You do not nominate
a complying superannuation fund for this purpose, the contribution may be paid into an existing stapled account identified by
the Commissioner of Taxation.
5.2Annualised salary
Your base salary absorbs all entitlements, benefits and payments that You may now have or subsequently acquire. Your
base salary includes compensation for all entitlements, benefits or payments that might otherwise be due under the Fair
Work Act 2009 (Cth) (FW Act) or any industrial instrument that may apply to Your employment including overtime and
penalty payments for out of hours work and working weekends and public holidays, and any other loadings, penalties,
overtime or allowances. Accordingly, You will not be paid any special rates or allowances for working particular times or
under particular conditions unless otherwise agreed in writing. Further, any remuneration paid in excess of any particular
entitlement may be offset against any other entitlement, including an entitlement in a different pay period.
5.3Guarantee of annual earnings
The Company undertakes that the base salary and any other benefits paid to you is a guarantee of annual earnings for the
purposes of the FW Act for a period of at least 12 months. You accept that undertaking and acknowledge that Your
employment will not be subject to the application of any modern award for any period during which you earn in excess of
the high income threshold as defined by the FW Act.
5.4Payment
The Company will pay any Total Remuneration that You elect to receive as salary (less any compulsory superannuation
contributions and applicable tax), based on the Payment Frequency set out in the Offer of Employment, by electronic
transfer to Your nominated bank account.
5.5Expenses
The Company will reimburse You for any expenses that You reasonably incur during the performance of Your duties. The
Company may require You to provide a tax invoice, or other evidence, to substantiate any expenses claim.
6INCENTIVE ARRANGEMENTS
6.1Long Term Incentives
(a)You may be eligible to participate in long-term incentive arrangements offered by the Company from time to time.
(b)Your participation is subject to the rules of the plan. The Company may amend or discontinue the plan and the rules of the plan
from time to time. They do not form part of Your employment agreement.
(c)Any payment made to You under the plan will not give rise to an expectation or create any precedent for the awarding of any
subsequent payment(s).
(d)Your participation in the plan, and all benefits under the plan, are at the absolute discretion of the Company. Any payment to
You will be net of any applicable tax and superannuation contribution which the Company is required to make in respect of any
incentive payment.
(e)Unless otherwise agreed in writing, You are only entitled to receive a benefit under the plan if You are employed by the
Company and not subject to a notice of termination period on the date that the benefit is paid or provided.
6.2Short Term Incentives
(a)You may be eligible to participate in any short-term incentive plans of the Board, which may be determined by the Board from
time to time.
(b)You may be entitled to a bonus incentive of up to a portion of your Base Salary set out in the Offer of Employment, which, if
payable, will be payable based on Your performance against agreed performance milestones and may be paid annually in cash
or equity and at the Board’s discretion.
(c)Your eligibility and any payment is subject to the rules of any plan or policy in place from time to time. The Board may amend or
discontinue the plan and the rules of the plan from time to time. They do not form part of Your employment agreement.
(d)Any payment made to You under the plan will not give rise to an expectation or create any precedent for the awarding of any
subsequent payment(s).
(e)Your participation in the plan, and all benefits under the plan, are at the absolute discretion of the Board. Any payment to You
will be net of any applicable tax and superannuation contribution which the Company is required to make in respect of any
incentive payment.
(f)Unless otherwise agreed in writing, You are only entitled to receive a benefit under the plan if You are employed by the
Company and not subject to a notice of termination period on the date that the benefit is paid or provided.
7LEAVE
7.1Your entitlement
(a)You are entitled to paid and unpaid leave including annual leave, personal/carer’s leave, compassionate leave, family and
domestic violence leave, community service leave and parental leave in accordance with legislation.
(b)Currently, Your entitlements under the National Employment Standards are 4 weeks annual leave each year, 10 days paid personal/
carer’s leave each year (in certain circumstances You may also be able to access unpaid carer’s leave), compassionate leave,
and up to 12 months unpaid parental leave where You have responsibility for the care of a child (in certain circumstances You may
be able to extend this leave).
(c)Entitlements for part-time employees are pro-rated accordingly.
You must notify your Manager immediately before taking personal/carer's leave so that the Company may accommodate
your absence. If this is not possible, You must contact your Manager or the Company as soon as you are able to. The
Company may require you to provide a medical certificate or attend a medical examination at any time.
7.2Long service leave
You will be entitled to long service leave in accordance with applicable legislation.
7.3Public holidays
You will usually be entitled to paid leave on days declared as public holidays in the state or territory in which You work.
From time to time You may be required, and You will not reasonably refuse, to work on a public holiday in order to
perform Your duties. Your remuneration includes an amount to compensate for this.
8ENDING YOUR EMPLOYMENT
8.1Ending Your employment with notice
After the Probation Period and subject to clause 8.6:
(a)You may end Your employment at any time by giving the Employee Notice Period set out in the Offer of Employment; or
(b)the Company may end Your employment at any time by giving the Employer Notice Period set out in the Offer of Employment.
8.2Payment in lieu of notice period
In the event that either You or the Company provide notice in accordance with clause 8.1 above, the Company may elect
to:
(a)pay You in lieu of Your notice period; or
(b)require You to work for part of Your notice period and pay You in lieu of the balance of the period.
8.3Calculation of payments if Your employment ends
If Your employment ends, the Company will calculate any payments in lieu of notice or accrued leave in accordance with
applicable law.
8.4Deduction of amounts owed
(a)To the extent permitted by law, any outstanding advances or other payments due to the Company by You will be deducted
before payment of any amounts under this clause 8 are made to You.
(b)If the amounts owed by You to the Company at the date Your employment ends exceed amounts payable to You under this
clause 8, You agree to repay such amounts to the Company within 14 days of the date Your employment ends.
8.5Duties during notice period
If You or the Company gives notice ending Your employment, the Company may direct You at any time during the notice
period:
(a)not to attend work;
(b)not to perform all or part of Your duties;
(c)to perform duties which are different to Your usual duties, provided that You have the necessary skills and competencies to
perform these duties;
(d)to assist the Company with a proper hand over of the duties of the Position including business information, work, clients, and
business;
(e)not to have any dealings with any customers, suppliers or clients of the Company or the Group; or
(f)to do any combination of the above.
8.6Ending Your employment without notice
The Company may end Your employment at any time without notice if You:
(a)engage in serious or wilful misconduct;
(b)are seriously negligent in the performance of Your duties;
(c)commit a serious or persistent breach of Company policy (including but not limited to a serious or persistent beach of the Code
of Conduct);
(d)commit a serious or persistent breach of this agreement;
(e)refuse to carry out lawful and reasonable instructions of the Company;
(f)commit an act, whether at work or otherwise, which brings the Company into disrepute; or
(g)are charged with an offence punishable by imprisonment.
8.7Suspension
If the Company suspects that You have been involved in any improper conduct or involved in any other conduct which in
the opinion of the Company may impact upon Your ability to carry out Your duties and responsibilities under this
agreement or may cause damage to the Company's business or reputation, the Company may do any or all of the
following:
(a)suspend You from performing the duties and responsibilities of the Position for a period determined by the Company;
(b)direct You not to attend the workplace, communicate with fellow employees, customers, suppliers or clients of the Company or
any other persons involved in the conduct which is being investigated, or otherwise interfere with the conduct of the
investigation; and
(c)appoint any person to conduct the investigation; and direct You to provide any assistance and answer any questions required
for the investigation.
During the period of suspension, You will continue to receive Your Total Remuneration under this agreement. Any
suspension under this clause will not be treated as disciplinary action by the Company, but will be instituted solely for the
purpose of conducting an investigation.
8.8Return of property
Before Your employment ends, or as soon as practicable after it ends, You must return all property belonging to the
Company.
8.9Redundancy
If Your employment ends for a reason including that Your position is redundant, any payment to You on ending of Your
employment (other than a payment in lieu of Your minimum statutory entitlement to notice of termination) will be inclusive
of any statutory entitlement that You may have to redundancy pay.
8.10Consideration for holding office
You and the Company agree that the benefits to which You are entitled under this agreement in connection with the
termination of Your employment are in part consideration for You agreeing to accept the office of the Position, and any
other office or position that is a ‘managerial or executive office’ (as that expression is defined in the Corporations Act
2001 (Cth)) in any Group company.
9DISCLOSURE OF INFORMATION
9.1Definitions
In this agreement:
(a)‘Confidential Information’ means any Information which is confidential and not in the public domain (unless in the public
domain because of a breach of confidentiality) including, but not limited to:
(i)the business or affairs, financial information, Intellectual Property, and sales and marketing information of the
Company or Group, or their respective customers or suppliers;
(ii)information which is marked “confidential” or which is described or treated by the Company or Group as confidential;
(iii)information of a business sensitive nature; and
(iv)trade secrets, research and confidential information and know-how of the Company, the Group, or their respective
customers or suppliers.
(b)‘Information’ means any information about the Group or its business (including, but not limited to, any idea, concept, process
or know-how) which comes to Your notice in the course of Your employment or is generated by You in the course of performing
Your duties.
9.2Importance of Confidential Information
(a)During Your employment with the Company You will acquire experience, Confidential Information, trade secrets, know-how and
particular skills in the affairs, practices, customer requirements and trade connections of the Group.
(b)Because of the importance to the Group of the knowledge and Confidential Information which You will acquire, the Company
wishes to ensure that You do not take advantage of these matters for Your advantage or others to the detriment of the Group
and its businesses and in violation of its rights.
9.3Your obligations during employment
During Your employment, You must not use or disclose Information unless the use or disclosure is:
(a)required by law;
(b)made as part of the proper performance of Your duties; or
(c)agreed in writing by the Company.
9.4Your obligations after Your employment ends
After Your employment ends, and without limiting Your general law obligations, You must not disclose Confidential
Information unless the disclosure is:
(a)required by law; or
(b)agreed in writing by the Company.
9.5Preventing disclosure
You must take all reasonable and necessary precautions to maintain the secrecy and prevent disclosure of Information.
You must immediately notify the Company if You suspect or become aware that Confidential Information has been
improperly copied, used or disclosed.
9.6Implied term and survival of obligations
(a)To avoid doubt, this clause is not intended to limit any duty of fidelity owed by You and implied into Your employment
agreement.
(b)Your obligations under this clause continue after Your employment ends.
10INTELLECTUAL PROPERTY
10.1Definition
In this clause ‘Intellectual Property’ means all present and future rights to intellectual property including any inventions and
improvements, trade marks (whether registered or common law trade marks), designs, copyright, patents, any
corresponding property rights under the laws of any jurisdiction and any rights in respect of any work, including any
invention, discovery, trade secret, secret process, know-how, concept, idea, information, process, data or formula.
10.2Ownership
(a)The Company owns all Intellectual Property that You create or contribute to during Your employment.
(b)You must do all things necessary to ensure that the Company owns Intellectual Property that You create or contribute to during
Your employment.
10.3Disclosure
You must inform the Company of all Intellectual Property that You create or contribute to during Your employment.
10.4Survival of obligations
Your obligations under this clause continue after Your employment ends.
11MORAL RIGHTS
(a)In this clause ‘Moral Rights’ means the right of attribution of authorship, the right not to have authorship falsely attributed and
the right of integrity of authorship, as defined in the Copyright Act 1968 (Cth).
(b)If You have Moral Rights in any Intellectual Property owned by the Company, You irrevocably consent to any act or omission
by the Company which infringes those Moral Rights. You agree that Your consent extends to acts and omissions by the
Company’s licensees and successors in title, and You agree that Your consent is a genuine consent given under Part 9 of the
Copyright Act 1968 (Cth) and has not been induced by duress or any false or misleading statement.
(c)Your obligations under this clause continue after Your employment ends.
12RESTRICTIONS DURING YOUR EMPLOYMENT
12.1Other business interests
Subject to clause 12.2, during Your employment You must not be engaged, concerned or interested in any other business
without the Company’s prior written consent, with such consent not to be unreasonably withheld if such business is not in
conflict with clause 2.3 or if disclosed to the Company prior to the commencement of Your employment.
12.2Shareholding
Despite any other clause of this agreement, You may hold shares in companies listed on any recognised stock exchange
without the Company’s prior written consent if You hold less than 5% of the issued shares of any class of any one
company.
13RESTRICTIONS AFTER YOUR EMPLOYMENT ENDS
13.1Restrictions
The Post Employment Restraints set out in the Offer of Employment apply to You.
13.2Consent
The Post Employment Restraints do not apply in circumstances where You have obtained the Company’s prior written
consent.
13.3Restrictions reasonable and independent
You agree that:
(a)You will obtain Confidential Information during Your employment, the disclosure of which could materially harm the Group;
(b)You will develop influence over customers, directors, employees and/or contractors of the Group;
(c)the Post Employment Restraints are reasonable and necessary for the protection of the Group’s Confidential Information,
goodwill and relationships with its customers, directors, employees and contractors;
(d)You intend the restrictions to operate to the maximum extent;
(e)damages may be inadequate to protect the Group’s interests and the Group is entitled to seek and obtain injunctive relief, or any
other remedy, in any court; and
(f)the restrictions are separate, distinct and several, so that the unenforceability of any restriction does not affect the
enforceability of the other restrictions.
13.4Modification of restrictions
If the Post Employment Restraints:
(a)are void as unreasonable for the protection of the Group’s interests; and
(b)would be valid if part of the wording was deleted or the period or area was reduced,
the restrictions will apply with the modifications necessary to make them effective.
13.5Obligations continue
Your obligations under the Post Employment Restraints and this clause 13 survive the ending of Your employment.
14COMPLIANCE AND APPROVALS
(a)The exercise of, or compliance with, any discretion, right or obligation under this agreement is subject to any required board or
shareholder approvals, any necessary regulatory consent and compliance with the Company’s constitution and all applicable
laws.
(b)Notwithstanding any provision of this agreement, the Company is not required to pay or provide, or procure the payment or
provision, of any payment or benefit to You which is not permitted by the provisions of Part 2D.2, Division 2 or Chapter 2E of
the Corporations Act 2001 (Cth) in the absence of shareholder approval. Any such payments or benefits must be reduced to
ensure compliance with this clause and there is no obligation on the Company to seek or obtain shareholder approval. In the
event of overpayment, You must, on receiving written notice from the Company Secretary (or his or her nominee), immediately
repay any monies or benefits specified in such notice.
(c)This clause 14 has effect regardless of any other provision of this agreement.
15GENERAL
15.1Governing law and jurisdiction
(a)This agreement is governed by the law in force in Governing law and jurisdiction in the Offer of Employment.
(b)Each party irrevocably submits to the non-exclusive jurisdiction of courts of the jurisdiction specified in the Offer of
Employment .
15.2Entire agreement and no reliance
(a)This agreement states all the express terms of the agreement between the parties in respect of its subject matter. It supersedes
all prior discussions, negotiations, understandings and agreements in respect of its subject matter.
(b)You acknowledge that in accepting employment with the Company You have not relied on any representations regarding Your
employment made by the Company (or its agents or employees) other than matters expressly set out in this agreement.
(c)This agreement may only be amended by agreement in writing signed by both parties.
15.3Legal advice
You represent that You have taken, or had the opportunity of taking, legal advice in relation to the nature, effect and
extent of this agreement.
15.4Counterparts
This agreement may be executed in any number of counterparts and all counterparts, taken together, constitute one
instrument. A party may execute this agreement by executing any counterpart.
15.5Benefit of this agreement
The Company executes this agreement for the Group. You acknowledge and agree that each Group Company may
independently enforce the obligations given in their favour in this agreement against You in their own right.
15.6Interpretation
In this agreement:
(a)A reference to the ‘Group’ means the Company and each ‘related body corporate' as that expression is defined in the
Corporations Act 2001 (Cth).
(b)Headings and bold type are for convenience only and do not affect the interpretation of this agreement.
(c)The singular includes the plural and the plural includes the singular.
(d)Words of any gender include all genders.
(e)Other parts of speech and grammatical forms of a word or phrase defined in this agreement have a corresponding meaning.
(f)An expression importing a person includes any company, partnership, joint venture, association, corporation or other body
corporate and any government agency as well as an individual.
(g)A reference to a clause, party, schedule, attachment or exhibit is a reference to a clause of, and a party, schedule, attachment or
exhibit to, this agreement.
(h)A reference to any legislation includes all delegated legislation made under it and amendments, consolidations, replacements or
re-enactments of any of them.
(i)No provision of this agreement will be construed adversely to a party because that party was responsible for the preparation of
this agreement or that provision.
(j)Specifying anything in this agreement after the words ‘include’ or ‘for example’ or similar expressions does not limit what else is
included.
(k)This agreement includes any schedule.
SIGNING PAGE
Executed as an agreement
COMPANY
EXECUTED on behalf of:
TELIX PHARMACEUTICALS
(CORPORATE) PTY LTD by:
/s/ Christian Behrenbruch | Christian BEHRENBRUCH | 18-Dec-23 | ||
Signature of authorised representative /s/ Meredith Crowe | Name of authorised representative Meredith Crowe | Date 18-Dec-23 | ||
Signature of witness | Name of witness | Date |
YOU
EXECUTED by David Cade in the presence of:
/s/ David Cade19-Dec-23
Signature of David CadeDate


/s/ Sharon Brewster20-Dec-23
Signature of witnessName of witness (please print)
Telix Pharmaceuticals (Corporate) Pty Ltd
Registered in Australia under A.C.N. 666 576 343
Registered Address: Level 4, 55 Flemington Road,
North Melbourne, Victoria, 3051, Australia
www.telixpharma.com
Dec 18, 2023
Private and confidential
David Cade
[***]
Offer of Employment
Dear David,
We are pleased to offer you employment with Telix Pharmaceuticals, as set out below. Terms and conditions of this appointment
are conditional of acceptance of the attached Employment Agreement.
Key Details | |
1. Position Title | Group Chief Medical Officer |
2. Employment Type | Full-time |
3. Contract Type | Permanent |
4. Employment Dates | Start Date: Jan 01, 2024 |
5. Contracted Hours | 37.5 (1.0 FTE) |
6. Location | 7.01, 10 Bridge Street, Sydney NSW 2000 |
7. Key Activities | Refer to attached Position Description |
8. Manager | Managing Director & Group CEO |
9. Total Remuneration | (a)base salary of $490,000 AUD per annum; plus (b)compulsory superannuation contributions paid by the Company for Your benefit, but does not include any payroll tax or workers’ compensation insurance paid by the Company in respect of Your employment. |
10. Payment Frequency | Monthly |
11. Short Term Variable Remuneration (STVR) | You may be eligible to receive an STVR of up to 35% of your base salary, subject to the terms set out in the Employment Agreement. Whether the payment is made and the amount of any payment is in the absolute discretion of the Board and the Company. |
12. Long Term Variable Remuneration (LTVR) | At the discretion of Telix Pharmaceuticals and subject to all relevant terms from your original employment agreement and LTVR letters issued to you, your long-term incentive may be up to 60% of your base salary. |
13. Employee Notice Period | 4 months’ notice |
Telix Pharmaceuticals (Corporate) Pty Ltd
Registered in Australia under A.C.N. 666 576 343
Registered Address: Level 4, 55 Flemington Road,
North Melbourne, Victoria, 3051, Australia
www.telixpharma.com
Key Details | |
14. Employer Notice Period | 4 months’ notice |
15. Post Employment Restraints | (a)No competing with the Group Unless the Company provides prior written consent, You must not (whether directly or indirectly) during the Restricted Period (explained below) and in the Restricted Area (also explained below) carry on, be employed by or engaged or otherwise interested in any Competitive Business (being any business that competes with the Group during the period of 3 months preceding the End Date, or during the Restricted Period) to provide products or services which are the same as or similar to those You provided to the Company at any time within the 3 months immediately preceding the date on which Your employment ends. (b)No inducing employees or contractors to leave the Group You must not during the Restricted Period and in the Restricted Area induce or attempt to induce any director, employee or contractor of the Group with whom You had work related dealings during the 3 months preceding the date on which Your employment ends to terminate his or her employment or engagement with the Group, whether or not that person would commit a breach of that person’s contract of employment or engagement. (c)No persuading the Group’s customer to cease or reduce business You must not during the Restricted Period and in the Restricted Area solicit or persuade any customer of the Group with whom You had work related dealings during the 3 months preceding the date on which Your employment ends to cease doing business with the Group or reduce the amount of business which the person |
16. Restricted Area | (a)Australia; or if a Court finds that unenforceable (b)Melbourne, Victoria, Australia |
17. Restricted Period | The period of 6 months starting on the End Date; or if a Court finds that unenforceable. |
18. Governing law and jurisdiction | Victoria, Australia |
19. Review Date | Your Total Remuneration package will be reviewed annually as part of the Company's remuneration review process. In undertaking this review, the Company may have regard to any matter in its absolute discretion. This review will not necessarily lead to an increase in Your Total Remuneration. |
Telix Pharmaceuticals (Corporate) Pty Ltd
Registered in Australia under A.C.N. 666 576 343
Registered Address: Level 4, 55 Flemington Road,
North Melbourne, Victoria, 3051, Australia
www.telixpharma.com
We look forward to having you join us at Telix and contributing to our vision of helping people with cancer and rare disease live
longer, better quality lives.
Executed as an agreement
COMPANY
EXECUTED on behalf of:
TELIX PHARMACEUTICALS
(CORPORATE) PTY LTD by:
/s/ Christian Behrenbruch | Christian BEHRENBRUCH | 18-Dec-23 | ||
Signature of authorised representative /s/ Meredith Crowe | Name of authorised representative Meredith Crowe | Date 18-Dec-23 | ||
Signature of witness | Name of witness | Date |
YOU
EXECUTED by David Cade in the presence of:
/s/ David Cade19-Dec-23
Signature of David CadeDate
/s/ Sharon Brewster20-Dec-23
Signature of witnessName of witness (please print)
Exhibit 4.11
CERTAIN INFORMATION (INDICATED BY “[***]”) CONTAINED IN THIS AGREEMENT HAS BEEN REDACTED BECAUSE (I) THE
COMPANY CUSTOMARILY AND ACTUALLY TREATS THE REDACTED INFORMATION AS PRIVATE OR CONFIDENTIAL AND (II) THE
OMITTED INFORMATION IS NOT MATERIAL.
Telix Pharmaceuticals (US) Inc.
11700 Exit 5 Pkwy Suite 200
Fishers, IN, 46037
USA

3/5/2024
Darren Patti
[***]
Dear Darren,

.
On behalf of Telix Pharmaceuticals (US), Inc. (“Telix”), I am pleased to offer you a change in employment with our team, effective March
11, 2024, on the terms and conditions set forth in this letter (the “Offer Letter”).This Offer Letter is contingent on your satisfaction of the
terms set forth below, and you should not take any action in reliance on this offer until you have satisfied these terms.
The terms of employment set forth in this Offer Letter replace and supersede all prior agreements, understandings, promises or
contracts between you and Telix regarding your employment, including without limitation any prior offer letters, employment agreements,
emails, or letters to you from Telix representatives that predate this Offer Letter.
Position and Duties. Your job title and position will be Group Chief Operating Officer, a full time, exempt position reporting to:
Managing Director & Group CEO. Your primary responsibilities can be found in the attached position description. Telix may modify your
job title, work location, duties, and responsibilities from time to time as it deems necessary.
Base Salary. Your starting annual base salary will be $360,000.00 USD, payable in accordance with Telix’s normal payroll procedures.
Your salary and performance will be reviewed periodically, and your salary may be increased or decreased in connection with any such
review.
Long Term and Short Term Variable Remuneration. You will be eligible to participate in any Long Term and Short Term variable
remuneration opportunities, subject to the terms of such plans. You will be eligible to receive an annual short-term variable remuneration
(STVR) bonus payment of up to 35% of your base remuneration for performance against objectives. The STVR may be paid in cash or
equity at the Board’s discretion. You will be eligible for long-term variable remuneration equal to 60% of your base remuneration,
typically issued as Performance Share Appreciation Rights (PSARs). PSARs vest over a three-year period and are contingent on the
achievement of key commercial and corporate objectives. LTVR rights are granted annually as a standard component of executive pay.
Your participation in the plan, and all benefits under the plan, are at the absolute discretion of the Company and are subject to the rules
of the plan.
1

Telix Pharmaceuticals (US) Inc. 11700 Exit 5 Pkwy Suite 200 Fishers, IN, 46037 USA |
Paid Time Off. You will be eligible to receive 20 days of paid time off (“PTO”) each year and 5 paid sick days each year, in accordance
with Telix’s PTO policy in effect for your work location. This will be pro-rated based on your date of hire for your first year. Your eligibility and
entitlement to PTO benefits will be governed by the terms and conditions of the PTO policy, except that PTO benefits may be used for
any purpose and in the manner authorized by all paid sick leave laws or ordinances that apply to your employment. The PTO policy is
subject to change or discontinuation at any time.
Employee Benefit Plans. Telix currently maintains medical, dental and vision employee benefit plans and offers a 401(k) plan with an
employer match. During your employment, you will be eligible to participate in the employee benefit and insurance plans maintained by
Telix for similarly situated employees, subject to the terms and conditions of the plans, as in effect from time to time. You also will be
eligible to receive certain perquisites offered by Telix to employees, including fitness, transportation, cell phone expenses, and home
Internet. Please note that Telix’s employee perquisites and benefit plans are subject to change or discontinuation and that your
participation in each plan is governed by the specific terms of the plan. Additional information about our benefit plans will be provided to
you via separate cover.
At Will Employment. Your employment with Telix is and shall at all times be at-will, meaning that your employment is not guaranteed
for any specified time period, that either Telix or you may terminate your employment at any time for any reason, with or without cause,
and with or without advance notice, and that Telix may modify your job title, work location, duties and responsibilities from time to time
as it deems necessary. The at-will nature of your employment cannot be changed except through a writing signed by both you and
Telix’s Chief Executive Officer.
Withholding. Telix will withhold federal, state and local income, employment or other taxes as required by applicable law from all
compensation or benefits paid to you in connection with your employment.
Compliance with Laws & Company Policies. As an employee of Telix, you will be expected to comply with all laws applicable to the
performance of your duties and responsibilities to Telix. You will also be expected to comply with Telix’s personnel and other policies.
Confidential Information, Intellectual Property Assignment, and Restrictive Covenant Agreement. As a condition of your
employment with Telix, you must read, agree to, and sign the Confidential Information, Intellectual Property Assignment, and Restrictive
Covenant Agreement (the “Confidentiality Agreement”) enclosed with this Offer Letter.
2
Telix Pharmaceuticals (US) Inc. 11700 Exit 5 Pkwy Suite 200 Fishers, IN, 46037 USA |
Entire Agreement; No Other Promises. This Offer Letter, in conjunction with the Confidentiality Agreement, and the Joining incentive
letter, contain the entire agreement between you and Telix concerning the terms and conditions of employment and replaces,
supersedes, and cancels all prior agreements, commitments, and understandings, whether spoken or written, that Telix may have made
in connection with your employment. No commitments affecting the terms of your employment or altering your employment status are
binding on Telix unless contained in a writing signed by both you and Telix’s Chief Executive Officer. You also acknowledge that the
agreement concerning the terms of your employment set forth in this Offer Letter is intended as written, and that no marginal notations
or other revisions to this Offer Letter, or the Confidentiality Agreement, are binding on Telix unless Telix’s Chief Executive Officer
expressly consents in writing to the revision. You acknowledge that in deciding to accept employment with Telix after the date of this
Offer Letter, you have not relied on any promises, commitments, statements, or representations, whether spoken or in writing, made to
you by any Telix representative, except for what is expressly stated in this letter and in the Confidentiality Agreement.
Other Terms. Any dispute arising out of or relating to this Letter or your employment with Telix shall be construed, governed by and
enforced in accordance with the laws of the state of Indiana or jurisdiction in which you primarily rendered services as an employee of
Telix, without giving effect to principles of conflicts of laws.
We look forward to our employment relationship with you. Please sign and date this letter below to indicate your acknowledgement and
understanding of the terms contained in this letter within 3 days of receipt. The offer of employment will expire if not accepted by that
time. If you have any questions, please contact your Talent Acquisition Representative.
3
Telix Pharmaceuticals (US) Inc. 11700 Exit 5 Pkwy Suite 200 Fishers, IN, 46037 USA |
Signing Page

Executed as an agreement
COMPANY
EXECUTED by
TELIX PHARMACEUTICALS (US) INC. by:
Kris King
/s/ Kris King
Telix Pharmaceuticals (US) Inc. 11700 Exit 5 Pkwy Suite 200 Fishers, IN, 46037 USA |
Signature of People & Culture
Representative
Director, P&C Americas
Chris Behrenbruch
/s/ Chris Behrenbruch
Signature of Hiring ManagerManaging Director & Group CEO
YOU
EXECUTED by:
/s/ Darren Patti
Darren Patti
CONFIDENTIAL INFORMATION, INTELLECTUAL PROPERTY ASSIGNMENT, AND RESTRICTIVE COVENANT AGREEMENT
As a condition of my employment with Telix Pharmaceuticals (US) Inc. (the “Company” or “Employer”), and in consideration of my
employment with the Company and my receipt of the compensation and benefits paid to me by the Company, I agree to the terms and
conditions of this Confidential Information, Intellectual Property Assignment, and Restrictive Covenant Agreement (the “Confidentiality
Agreement”). In this course of my employment, I will be provided with and learn confidential information regarding the Company’s (as
defined below in Section 1), and its customers, and/or will establish, maintain and improve knowledge of or relationships or goodwill with
the Company’s customers, or will learn the Company’s Trade Secrets or Confidential Information (as such terms are defined below); I
acknowledge the Company will not employ or may not continue to employ me in my current position if I do not accept the terms
outlined herein:
4
Telix Pharmaceuticals (US) Inc.
11700 Exit 5 Pkwy Suite 200
Fishers, IN, 46037
USA
1.Non-Disclosure and Non-Use of Confidential Information and Ownership of Intellectual Property.
(a)I acknowledge that, during the course of my employment, I will have access to information about the Company and its
respective parent companies and direct and indirect subsidiaries and affiliates (collectively, the “Company Group”) and that my
employment with the Company shall bring me into close contact with confidential and proprietary information of the Company Group. In
recognition of the foregoing, I agree, at all times during the term of my employment with the Company (the “Employment Period”) and for
three years after my Termination Date to hold in confidence, and not to use, except for the benefit of the Company Group, or to disclose
to any Person (as defined below) except as required in the performance of my authorized duties to the Company or with written
authorization of the Company, any Confidential Information that I obtain or create in the course of my employment. I further agree not to
make copies of such Confidential Information except as required in the performance of my authorized duties to the Company or as
authorized by the Company. I understand that “Confidential Information” means all information heretofore or hereafter developed or used
by the Company Group (whether or not reduced to written, electronic, magnetic or other tangible form) to which I had access during the
course of my employment with the Company Group and which is proprietary to the Company Group and not disclosed to the public by
the Company Group in the ordinary course of its business or which relates to any third party for which the Company Group is under an
obligation to keep such information confidential, concerning the research, product development, products, operations, marketing and
business plans, activities, consultants, licensors, licensees, customers, or business affairs of the Company Group, or the Company
Group’s licensees, distributors, business partners or customers, including, without limitation: (A) all information concerning Trade
Secrets of the Company Group, including data lists, directories, computer programs, system documentation, special hardware, product
hardware, related software development, computer systems, source code, object code, manuals, formulae, processes, methods,
machines, compositions, ideas, improvements or inventions; (B) all sales and financial information concerning the Company Group; (C)
all customer information, customer lists, or customer preferences or requirements; (D) all Company Group strategy, research activities,
data, technology, methodologies, techniques, distribution plans, contractual arrangements, profits, sales, price lists, pricing policies,
operational methods, technical processes, other business affairs and methods, plans for future developments and other technical and
business information relating to the business of the Company Group and their business partners or customers and all trademarks,
domain names, copyrights and patents and applications thereof, all inventions, processes, studies, reports, research records, market
surveys and know-how and technical papers; (E) all information in any way concerning the business or affairs of the Company Group’s
affiliates, suppliers, business partners or customers which was furnished to me by the Company Group, suppliers, business partners or
customers or otherwise discovered by me during my employment with the Company; and (F) any document marked “confidential” or any
information which I have been advised is confidential or which might reasonably be expected to be regarded as confidential or any
information which has been given to the Company Group in confidence by customers, suppliers or other persons. “Trade Secret” means
a Trade Secret as that term is defined under Illinois law and under the Economic Espionage of 1996 and the Defend Trade Secrets Act
of 2016, and their amendments. Notwithstanding the foregoing, Confidential Information shall not include (i) any of the foregoing items
that have become publicly and widely known through no unauthorized disclosure by me or others who were under confidentiality
obligations as to the item or items involved or (ii) any information that I am required to disclose to, or by, any governmental or judicial
authority; provided, however, that in such event I will give the Company prompt written notice thereof so that the Company Group may
seek an appropriate protective order and/or waive in writing compliance with the confidentiality provisions of this Confidentiality
Agreement.
5
Telix Pharmaceuticals (US) Inc. 11700 Exit 5 Pkwy Suite 200 Fishers, IN, 46037 USA |
(b)Nothing in this Confidentiality Agreement shall prohibit or impede me (or my attorney) from responding to any
inquiry about the Agreement or its underlying facts and circumstances by a U.S. federal, state or local governmental or law
enforcement branch, agency or entity (collectively, a “Governmental Entity”), or making other disclosures that are protected under
the whistleblower provisions of federal or state law or regulation. In addition, nothing in this Confidentiality Agreement prohibits me from
reporting possible violations of federal, state or local law to any governmental agency or entity. I understand and acknowledge that I do not
need the prior authorization of the Company to make any such reports or disclosures and that I am not required to notify the
Company that I have made such reports or disclosures. In addition, nothing in this Confidentiality Agreement is intended to interfere
with any rights I may have under Section 7 of the National Labor Relations Act.
(c)Notwithstanding anything in this Confidentiality Agreement to the contrary, I understand that I may, pursuant to the
U.S. Defend Trade Secrets Act of 2016 (“DTSA”), without informing the Company prior to any such disclosure, disclose
Confidential Information (i) in confidence to a federal, state, or local government official, either directly or indirectly, or to an attorney, solely
for the purpose of reporting or investigating a suspected violation of law or (ii) in a complaint or other document filed in a lawsuit or other
proceeding, if such filing is made under seal. Additionally, without informing the Company prior to any such disclosure, if I file a lawsuit
against the Company for retaliation for reporting a suspected violation of law, I may, pursuant to the DTSA, disclose Confidential
Information to my attorney and use the Confidential Information in the court proceeding or arbitration, provided that I file any document
containing the Confidential Information under seal and does not otherwise disclose the Confidential Information, except pursuant to court
order. Without prior authorization of the Company, however, the Company does not authorize me to disclose to any third party (including
any government official or any attorney I may retain) any communications that are covered by the Company’s attorney-client privilege.
6
Telix Pharmaceuticals (US) Inc. 11700 Exit 5 Pkwy Suite 200 Fishers, IN, 46037 USA |
2.Assignment of Intellectual Property
I agree to assign, and [as provided under applicable law], I hereby assign, to the Company irrevocably and unconditionally, all of my
rights, title, and interest in and to (1) any and all technology, discoveries, inventions and improvements, whether patentable or not,
creations, developments, reductions to practice, designs, processes, methods, techniques, practices, works of authorship and other
works, whether copyrightable or not, documentation, know-how, show-how, software, source code, object code, other code, systems,
data, database, devices, products, prototypes, specifications, applications, implementations, conceptions, ideas, and information in any
form and format (individually and collectively, “Intellectual Property”) made, developed, acquired, obtained, conceived, or suggested,
whether by me alone or with other(s), directly or indirectly, (i) relating to the Company’s business, whether existing, coming into
existence, or contemplated, or the Company’s actual or demonstrably anticipated research or development (whether during or after the
end of my employment with or engagement by the Company), or (ii) in the course of, as a result of, during the hours or time of, or in
connection with my employment or engagement by, or other performance for, the Company, or (iii) based on, including, resulting from, or
with the use of any Intellectual Property, or any equipment, supplies, tools, materials, or other personal property, funds, other resources,
facility, or location, belonging to, owned by, or provided, made available or accessible, or obtained or received from the Company
(whether during or after the end of my employment with or engagement by the Company), or (iv) is any derivative work, improvement,
modification, adaptation, translation, transliteration, or derivative of any Property owned or licensed by the Company (whether during or
after the end of my employment with or engagement by the Company) (all collectively and each individually, “Resulting Property”) and (2)
any and all patents, patent applications, patent rights, and utility models, all copyrights, all mask work rights, all trade secrets, all
trademarks, service marks, trade dress, trade names, and domain names, and all goodwill related thereto, all database rights, and all
other intellectual property rights, whether recognized now or in the future, and any registration or application for any of the foregoing, and
all rights and remedies related to any infringement or misappropriation of any of the foregoing, whether based on any past, present, or
future event, all anywhere in the world, whether existing now or coming into existence in the future (“IP Rights”) in and to any Resulting
Property (“Resulting IP Rights”). I will provide to the Company any Resulting Property promptly after making, developing, acquiring, or
conception such Resulting Property, and at any time upon the Company’s request, during or after my employment with the Company.
This includes, but is not limited to, providing all documentation, material, and files including or manifesting the Resulting Property. For
the avoidance of doubt, and as required by applicable law , I am not required to assign or offer to assign to the Company any of my
rights in an invention for which no equipment, supplies, facilities, or trade secret information of the Company was used and which was
developed entirely on my own time, unless (aa) the invention relates (I) to the business of the Company, or (II) to the Company’s actual
or demonstrably anticipated research or development, or (bb) the invention results from any work I performed for the Company. To the
extent that any Resulting Property includes, is based on, or derivative of any Property of mine not assigned to the Company, or that I
have any IP Rights not assigned to the Company, I hereby grant to the Company a non-exclusive, perpetual, non-terminable, irrevocable,
worldwide right and license, free of any royalty, fee, or other payment or payment obligation, to use, utilize, reproduce, distribute, create
derivative works, improvements, and derivations from, display, perform, and exploit such Property as part of or related to such Resulting
Property, and under all of my current and future IP Rights, to use, utilize, reproduce, distribute, create derivative works, improvements,
and derivations from, display, perform, and exploit any Resulting Property, which right and license is directly and indirectly sublicensable
and is assignable and transferable in connection with any license, assignment, or transfer of any Resulting Property or any Resulting IP
Rights. Without the Company’s express prior written consent, I shall not include any Property of a third party, or base on or derive from
any Property of a third party, any Resulting Property. Upon and as requested by the Company, I will execute or cause to be executed
such documents and agreements and take such other action (including, without limitation, providing any information and documents,
executing any documents and affidavits, providing any testimony, and/or rendering any other assistance) as may be desirable in the
Company’s opinion to effect any assignment and grant under this Section 2, or to file any application for, prosecute, secure, perfect, and
obtain registrations for any Resulting IP Rights, or to otherwise fully effect and implement the provisions in this Section 2. All of my
assignments, grants, obligations, and performance under this Section 2 is in consideration of my compensation and benefits provided
by the Company. For the avoidance of doubt, the Company has the sole right, as decided in its discretion, to exercise, enforce, and
exploit any Resulting IP Rights and use, utilize, reproduce, distribute, create derivative works, improvements, and derivations from,
display, perform, and exploit any Resulting Property, without any payment or obligation to pay any royalty, fee, or other amount or value
related thereto. The Company solely owns and retains, and does not assign, transfer, convey, or grant, expressly or implicitly, any right,
license, lien, or claim to me, in or to or under any right, title, or interest in or to any Resulting Property, any Resulting IP Rights, or any
other Property or IP Rights owned, licensed, or belonging to the Company or any part of the Company Group.
7
Telix Pharmaceuticals (US) Inc. 11700 Exit 5 Pkwy Suite 200 Fishers, IN, 46037 USA |
3.Restrictive Covenants and Duty of Loyalty.
(a)Non-Solicitation of Customers. During the Employment Period and the Restricted Period, I shall not, directly or
indirectly for my own account or for the account of any other Person (other than for the benefit of the Company Group), from any
location, directly or indirectly, for my own benefit or for the benefit of any other person, company, business entity or other
organization, for pay or otherwise, solicit, serve, or participate in soliciting or serving, any customer or prospective customer:
(i)with whom I had contact or dealings on behalf of the Company Group during the two (2) years immediately preceding
the Termination Date; or
(ii)for whom I was in a customer management capacity on behalf of the Company Group; or
8
Telix Pharmaceuticals (US) Inc.
11700 Exit 5 Pkwy Suite 200
Fishers, IN, 46037
USA
(iii)about which I learned Confidential Information, in each case for the purpose of competing with the Company or
Company Group in the Restricted Area.
(b)Non-Solicitation of Employees. During the Employment Period and the Restricted Period, I shall not, directly or
indirectly, for my own benefit or for the benefit of any other person, whether as an owner, director, officer, employee, agent, consultant, or
in any other capacity, whether for pay or otherwise:
(i)induce, solicit, entice or procure, any current or former Company employee to leave the employment of the Company
or the Company Group, where that person is an employee of the Company or the Company Group on the Termination Date; or
(ii)be personally involved to a material extent in: (i) accepting into employment or (ii) otherwise engaging or using the
services of, any person who is an employee of the Company or an affiliate of the Company on the Termination Date.
(c)Non-Competition
(i)During the Employment Period, I shall not directly or indirectly, alone or in association with or on behalf of any other
Person, carry on, be employed by, or engaged or otherwise interested in, for a Competitive Business.
(ii)During the Restricted Period, I shall not, within the Restricted Area, directly or indirectly, alone or in association with
or on behalf of any other Person; (A) carry on; (B) be employed by; (C) be engaged or otherwise interested in; or (D) perform or
provide services for a Competitive Business.
(d)Duty of Loyalty.
(i)During employment with the Company, I shall owe the Company an undivided duty of loyalty and shall take no action
adverse to that duty of loyalty. My duty of loyalty to the Company includes a duty to promptly disclose to the Company any
information that might cause the Company to take or refrain from taking any action or which otherwise might cause the
Company to alter its behavior.
(ii)I acknowledge that, in the event of an end of my employment with the Company, for any reason and at any time, I
will be able to earn a livelihood without violating the provisions of this Agreement. The Company and I acknowledge that my
rights have been limited only to the extent reasonably necessary to protect the Company’s legitimate interests. However, if I
believe in good faith that the restrictions in this Agreement will prevent me from obtaining a new job, I may notify the Company
in writing, providing reasonable details about the proposed responsibilities of the new job (without disclosing another person’s
confidential information). I will discuss with the Company whether appropriate accommodations can be made to protect the
Company’s interests while allowing me to take the new job. The Company shall be under no obligation to modify the restrictions
in this Agreement, but may do so in its sole and absolute discretion. Without limiting the generality of the foregoing, I shall
provide at least four (4) weeks written notice to the Company at any time that I decide to (a) terminate employment with the
Company or (b) enter into competition with the Company or (c) to enter into competition with Company Group where I had
access to Confidential Information, Trade Secrets or customer relationships. The Company may decide at such time to limit,
suspend, or terminate my employment or access to Confidential Information or customer relationships. If for any reason I
cannot, despite using my best efforts, provide four (4) weeks’ notice prior to accepting any such position, I agree to provide four
(4) weeks’ notice prior to commencing that new position. I acknowledge that a four (4) weeks’ notice period is appropriate and
necessary to permit the Company to determine whether, in its view, my proposed new position could lead to a violation of this
Agreement, and I agree to provide the Company with such information (except that I need not provide any information that would
constitute confidential or trade secret information of any third party). During the notice period required under this Section 3(d)(ii),
the Company may choose, in its sole discretion, to limit my duties in their position with the Company and to restrict my access
to the Company’s premises, systems and employees.
9
Telix Pharmaceuticals (US) Inc. 11700 Exit 5 Pkwy Suite 200 Fishers, IN, 46037 USA |
4.Definitions.
For purposes of this Confidentiality Agreement:
(a)“Competitive Business” means (i) any business involved in the development, pre- clinical or clinical, and/or the
commercialization of Radiopharmaceuticals for imaging or therapy for urologic and brain indications) to provide products or services
which are the same as or similar to those I provided to the Company at any time within the 6 months immediately preceding my
Termination Date; any business that develops, manufactures, produces, markets, sells or distributes any products or provides any
services of the kind (x) developed, under development, manufactured, produced, marketed, sold, distributed or provided by the Company
Group at the time of my Termination Date, or in which the Company Group was engaged during the two (2) years preceding my
Termination Date, or (y) in which the Company Group has plans to engage at the time of my Termination Date or (iii) otherwise engaging
in business activity that is competitive with products or services provided by the Company Group at any time during the twelve (12)
months preceding my Termination Date.
10
Telix Pharmaceuticals (US) Inc. 11700 Exit 5 Pkwy Suite 200 Fishers, IN, 46037 USA |
(b)“Person” shall mean any individual or natural person, partnership (including a limited liability partnership),
corporation, limited liability company, association, joint stock company, trust, joint venture, unincorporated organization or
governmental authority, who is not a party to this Confidentiality Agreement.
(c)“Restricted Area” means the greater of :(i) the United States of America; Australia, United Kingdom and the
European Union; (ii) states, provinces or territories within the United States of America or other countries in which I, or one or
more other Company employees or Company Group business units managed or directed by me (a) provided products or services on behalf
of the Company or Company Group; (b) sold or solicited the sale of products and services on behalf of the Company or Company Group; (c)
provided products or services designed, developed, tested or produced by me (either individually or in collaboration with other Company
Group employees), or by one or more other Company Group employees or business units managed by me in the twenty-four (24) month
period immediately preceding my Termination Date; or (iii) the United States of America.
(d)“Restricted Period” means a period of six (6) months after my Termination Date. The Restricted Period will be
extended beyond six (6) months up to a maximum of twelve months if I have breached a fiduciary duty to the Company or have unlawfully
taken, physically or electronically, property belonging to the Company Group.
(e)“Termination Date” shall mean the date that my employment with the Company terminates.
5.Reasonableness of Restrictions.
I acknowledge and recognize the highly competitive nature of the Company Group’s business, that access to Confidential
Information renders me special and unique within the Company Group’s industry, and that I will have the opportunity to develop
substantial relationships with existing and prospective clients, accounts, customers, consultants, contractors, investors, and strategic
partners of the Company Group during the course of and as a result of my employment with the Employer. In light of the foregoing, I
recognize and acknowledge that the restrictions and limitations set forth in this Confidentiality Agreement are reasonable and valid in
geographical and temporal scope and in all other respects and are essential to protect the value of the business and assets of the
Company Group, and that the Company would not employ me but for my agreements herein. I further acknowledge that the restrictions
and limitations set forth in this Confidentiality Agreement will not materially interfere with my ability to earn a living following the
termination of my employment with the Employer and that my ability to earn a livelihood without violating such restrictions is a material
condition to my employment with the Employer.
11
Telix Pharmaceuticals (US) Inc. 11700 Exit 5 Pkwy Suite 200 Fishers, IN, 46037 USA |
6.Independence; Severability; Blue Pencil.
Each of the rights enumerated in this Confidentiality Agreement shall be independent of the others and shall be in addition to
and not in lieu of any other rights and remedies available to the Company Group at law or in equity. If any of the provisions of this
Confidentiality Agreement or any part of any of them is hereafter construed or adjudicated to be invalid or unenforceable, the same shall
not affect the remainder of this Confidentiality Agreement, which shall be given full effect without regard to the invalid portions. If any of
the covenants contained herein are held to be invalid or unenforceable because of the duration of such provisions or the area or scope
covered thereby, I agree that the court making such determination shall have the power to reduce the duration, scope, and/or area of
such provision to the maximum and/or broadest duration, scope, and/or area permissible by law, and in its reduced form said provision
shall then be enforceable.
7.Injunctive Relief.
I expressly acknowledge that any breach or threatened breach of any of the terms and/or conditions set forth in this
Confidentiality Agreement may result in substantial, continuing, and irreparable injury to the members of the Company Group. Therefore,
I hereby agree that, in addition to any other remedy that may be available to the Company, any member of the Company Group shall be
entitled to seek injunctive relief, specific performance, or other equitable relief (without the requirement to post bond) by a court of
appropriate jurisdiction in the event of any breach or threatened breach of the terms of this Confidentiality Agreement without the
necessity of proving irreparable harm or injury as a result of such breach or threatened breach. Notwithstanding any other provision to
the contrary, I acknowledge and agree that the Post-Termination Restricted Period shall be tolled during any period of violation of any of
the covenants in Section 3 hereof and during any other period required for litigation during which the Company or any other member of
the Company Group seeks to enforce such covenants against me if it is ultimately determined that I was in breach of such covenants.
8.General Provisions.
(a)Governing Law; Jurisdiction. This Confidentiality Agreement is governed by the laws of the State of Illinois
without regard to its principles of conflict of laws. Any litigation regarding this Confidentiality Agreement must be brought in the
state courts or, if federal jurisdiction is appropriate, the federal courts of Illinois (collectively, the “Illinois Courts”). The parties agree that
jurisdiction and venue are proper in the Illinois Courts and waive any objection thereto.
(b)Waiver of Jury Trial. THE PARTIES WAIVE THEIR RIGHT TO A TRIAL BY JURY IN ANY LEGAL ACTION ARISING
OUT OF OR RELATING TO THIS CONFIDENTIALITY AGREEMENT AND MY OFFER LETTER.
(c)Entire Agreement. This Confidentiality Agreement and my Offer Letter set forth the entire agreement and
understanding between the Company and me relating to the subject matter herein and therein and merges all prior discussions between us.
No modification or amendment to this Confidentiality Agreement, nor any waiver of any rights under this Confidentiality Agreement, will be
effective unless in writing signed by the party to be charged. Any subsequent change or changes in my duties, obligations, rights, or
compensation will not affect the validity or scope of Confidentiality Agreement.
12
Telix Pharmaceuticals (US) Inc. 11700 Exit 5 Pkwy Suite 200 Fishers, IN, 46037 USA |
(d)Successors and Assigns. This Confidentiality Agreement will be binding upon my heirs, executors,
administrators, and other legal representatives and will be for the benefit of the Company, its successors, and its assigns. I
expressly acknowledge and agree that this Confidentiality Agreement may be assigned by the Company without my consent to any other
member of the Company Group as well as any purchaser of all or substantially all of the assets or stock of the Company or
Company Group, whether by purchase, merger, or other similar corporate transaction.
(e)Survival. The provisions of this Confidentiality Agreement shall survive the termination of my employment with the
Employer and/or the assignment of this Confidentiality Agreement by the Company to any successor in interest or other assignee.
* * * * *
[signature page follows]
13
Telix Pharmaceuticals (US) Inc. 11700 Exit 5 Pkwy Suite 200 Fishers, IN, 46037 USA |
IN WITNESS WHEREOF, the parties hereto have duly executed this Confidentiality Agreement as of the day and year first
above written.
Telix Pharmaceuticals (USA) Inc.
Date: March 5, 2024
Name:Kris King /s/ Kris King
Title: /s/ Director P&C - Americas
[EMPLOYEE]
/s/ Darren Patti
14Telix Pharmaceuticals (US) Inc.
Telix Pharmaceuticals (US) Inc. 11700 Exit 5 Pkwy Suite 200 Fishers, IN, 46037 USA |
APPENDIX A
This Appendix A modifies the Confidential Information, Intellectual Property Assignment, and Restrictive Covenant Agreement (the
“Confidentiality Agreement”) entered into between the Company and me, and is incorporated into and is part of that Confidentiality
Agreement.
California:
If I primarily reside and work for the Company in California, then:
(a) The post-employment restrictive covenants in Sections 3(a), 3(b), 3(c)(ii) and Section 3(d)(ii) shall not apply to me. However, conduct
involving misappropriation of Company trade secrets will remain prohibited and nothing in this Agreement shall be construed to limit or
eliminate any rights or remedies the Company would have against me under trade secret law, unfair competition law, or other laws
applicable in California absent this Agreement.
(b)No provision in this Agreement requires me to assign any of my rights to an invention if that invention qualifies for exclusion under
California Labor Code § 2870, which may be amended from time to time and which is incorporated by reference herein. The text of such
code states:
(i)Any provision in an employment agreement which provides that an employee shall assign, or offer to assign, any of
his or her rights in an invention to his or her employer shall not apply to an invention that the employee developed entirely
on his or her own time without using the employer’s equipment, supplies, facilities, or trade secret information except
for those inventions that either: (A) relate at the time of conception or reduction to practice of the invention to the employer’s
business, or actual or demonstrably anticipated research or development of the employer; or (B) result from any work
performed by the employee for the employer, (ii) to the extent a provision in an employment agreement purports to require
an employee to assign an invention otherwise excluded from being required to be assigned under subdivision (i), the
provision is against the public policy of this state and is unenforceable.
(c)All disputes arising from my employment or this Agreement shall be adjudicated in California.
15Telix Pharmaceuticals (US) Inc.
Telix Pharmaceuticals (US) Inc. 11700 Exit 5 Pkwy Suite 200 Fishers, IN, 46037 USA |
Colorado:
If my relationship with the Company is such that a court of competent jurisdiction would apply the law of Colorado to interpret this
Agreement: If I do not qualify as executive or management personnel, an officer, or an employee who constitutes professional staff to
executive and management personnel within the meaning of § 8-2-113(2)(d) of Colorado Revised Statutes § 8-2-113, et. seq. (the
“Colorado Noncompete Act”), its successor statutes and interpretive case law, then: the post-employment restrictions in Sections 3(a),
3(b), and 3(c) will not apply to me. However, conduct involving misappropriation of Company trade secrets or Confidential Information will
remain prohibited as provided in the Confidentiality Agreement and nothing in this Confidentiality Agreement shall be construed to limit
or eliminate any rights or remedies the Company would have against me under trade secret law, unfair competition law, or other laws
applicable in Colorado.
Florida:
If Florida law controls, then: I acknowledge and understand that my employment with the Company gives me access to and knowledge
of Confidential Information and places me in a position of trust and confidence with the Company Group. I also acknowledge and
understand that the Company Group’s ability to reserve its Confidential Information for the exclusive knowledge and use of the Company
Group is of great competitive importance and commercial value to the Company Group, and that improper use or disclosure of this
information by me is likely to result in unfair or unlawful competitive activity. I further acknowledge and understand that the restrictive
covenants in this Agreement are necessary to protect these legitimate business interests of the Company Group.
Georgia:
If Georgia law controls then: The definition of the Restricted Area referred to in the Agreement shall be understood to be the territory
where I (the employee) am working at the time of termination and I stipulate that the provisions of the Agreement provide me with
adequate means to reasonably determine the maximum scope of the restraints placed upon me at the time of my employment
termination. The definition of Confidential Information shall exclude data or information (A) which has been voluntarily disclosed to the
public by the Company, except where such public disclosure has been made by me or another employee without authorization from the
Company; (B) which has been independently developed and disclosed by others; or (C) which has otherwise entered the public domain
through lawful means.
Illinois:
If Illinois law controls then: The restrictive covenant in Section 3(c) will not apply to me if I am paid $13.00 per hour or less.
16Telix Pharmaceuticals (US) Inc.
Telix Pharmaceuticals (US) Inc. 11700 Exit 5 Pkwy Suite 200 Fishers, IN, 46037 USA |
Massachusetts:
If Massachusetts law controls then: If I breach the non-compete covenant in Section 3(c) and also breach my fiduciary duty to the
Company and/or I have unlawfully taken, physically or electronically, any Company Confidential Information, then the Restricted Period
shall be extended to a period of two (2) years from the Termination Date. Further, the covenant in Section 3(c) will not apply to me if my
employment is terminated without cause or if I am terminated as part of a reduction in force. For purposes of the foregoing test only,
“cause” to terminate my employment will exist if the Company concludes I have (i) committed, admitted committing, or plead guilty to a
felony or crime involving moral turpitude, fraud, theft, misappropriation, or dishonesty, (ii) violated a material term of this Agreement or
Company policy, (iii) engaged in insubordination, or failed or refused to perform assigned duties of my position despite reasonable
opportunity to perform, (iv) failed to exercise reasonable care and diligence in the exercise my duties for the Company, or (iv) engaged in
conduct or omissions that I knew, or should have known (with the exercise of reasonable care), would cause, or be likely to cause, harm
to the Company or its reputation in the business community. If I am being initially hired by the Company I confirm that I received a copy
of this Agreement prior to receiving a formal offer of employment from the Company or at least ten (10) business days before
commencement of my employment, whichever came first; and if I was already employed by the Company at the time of signing this
Agreement, I confirm that I was provided a copy of this Agreement at least ten (10) business days before the effective date. I
acknowledge and agree that I have received fair and reasonable consideration in exchange for my post-employment non-competition
covenant. The covenant in Section 3(c) shall not apply to me post-employment if I am classified as non-exempt under the Fair Labor
Standards Act, 18 years or younger, or an undergraduate or graduate student in an internship or other short-term employment
relationship while enrolled in college or graduate school.
Texas:
If Texas law controls then: The restrictive covenant in Section 3(c) shall not apply to any Competitive Business with whom I had no
contact or dealings on behalf of the Company Group during the two (2) years immediately preceding the Termination Date.
Virginia:
If Virginia law controls then: The restrictive covenant in Section 3(c) will not apply to me if I am a low-wage employee, as that term is
defined in 40.1-28.7:8 of the Virginia Code.
17Telix Pharmaceuticals (US) Inc.
Telix Pharmaceuticals (US) Inc. 11700 Exit 5 Pkwy Suite 200 Fishers, IN, 46037 USA |
Washington:
If Washington law controls then: The post-employment restrictive covenant in Section 3(c) will not be or become enforceable against me
unless or until my earnings from the Company, when annualized, exceed one hundred thousand dollars per year ($100,000/yr) or the
then inflation-adjusted equivalent in accordance with the requirements of Washington Noncompete Act (Chapter of Title 49 RCW
enacting ESHB 1450 of the 66th Legislature, 2019 Regular Session) (the “Washington Act”). In the event my employment is terminated
as a result of a layoff, the post-employment restrictive covenant in Section 3(c) will not be enforced by the Company unless the
Company agrees at the time of my layoff to provide me with the payments required by the Washington Act to keep the obligation under
Section 3(c)(ii) in effect. I further confirm that I was given ten (10) business days to consider this Agreement before accepting it, and if I
am a newly hired employee, I was given advance notice of the terms of this Agreement prior to accepting the Company’s offer of
employment.
18Telix Pharmaceuticals (US) Inc.
Exhibit 4.19

Deed of Indemnity and Insurance
Telix Pharmaceuticals Limited
ACN 616 620 369
and
insert Officer name
Page | 2
TEL005_1700042_030.DOC
Table of Contents |
to update table of contents - press F9
Page | 3
TEL005_1700042_030.DOC
Deed of Indemnity and Insurance |
This deed is made on between the following parties:
Telix Pharmaceuticals Limited ACN 616 620 369 of 55 Flemington Road, North Melbourne VIC 3051 Australia (the Company); and |
[name] of [address] (the Officer). |
Background | (A) | The Officer was determined as being an officer of the Company or Relevant Company (as the case may be) as of the Appointment Date. | |
(B) | In consideration of the Officer agreeing to act in his or her capacity as an officer of the Company or Relevant Company (as the case may be), the Company agrees to: | ||
(i) | indemnify the Officer against Liabilities incurred while acting as officer of the Company or Relevant Company (as the case may be); and | ||
(ii) | maintain a D&O Policy, | ||
on the terms contained in this deed. | |||
This deed witnesses that in consideration of, among other things, the mutual promises contained in
this deed, the parties agree as follows:
1Definitions and Interpretation |
1.1Definitions
In this deed:
Defined term | Meaning |
Affiliate | has the meaning as defined under the 15 U.S Code. |
Appointment Date | the date the Officer was appointed an officer or key management personnel of the Company (as defined under AASB 124, in section 9 of the Corporations Act or other applicable laws or regulations), or as an officer of Relevant Company (as the case may be). |
Board | the board of directors of the Company. |
Business Day | a day other than a Saturday, Sunday, bank holiday or public holiday in Melbourne, Victoria. |
Claim | (a)any legal proceeding, administrative proceeding, arbitral proceeding, investigation or enquiry, mediation, or other form of alternative dispute resolution, arising out of or in connection with any act or omission by the Officer or otherwise involving the Officer in their capacity as an officer; and (b)any written or oral threat, complaint or demand or other circumstances that might reasonably lead to the Officer considering that any proceedings set out in paragraph (a) will be commenced. |
Constitution | the Company’s constitution as amended, varied or replaced from time to time. |
Page | 4
Defined term | Meaning |
Corporations Act | the Corporations Act 2001 (Cth). |
D&O Policy | a policy of insurance insuring the Directors and Officer (amongst others) against liability in their capacity as Director and/or officer of the Company and its Related Bodies Corporate. |
Group Entities | the Company and any Subsidiary or Affiliate of the Company. |
Liability | a liability of any kind (whether actual or contingent and whether fixed or ascertained) including costs, damages, fees, expenses, and including whether the costs and expenses are incurred in connection with any investigation or inquiry by a government agency or liquidator. |
officer | has the meaning given to it for the purposes of the Corporations Act. |
Permitted Purpose | (a)defending or responding to an action or proceeding (or preparing to defend an action or proceeding which the Officer has reason to believe will be brought against them) which relates to an act or omission of the Officer in providing services in their capacity as an officer of the Company or Relevant Company (as the case may be) during the Relevant Period; (b)appearing before an inquiry or hearing of a Regulatory Body (or preparing for an inquiry or hearing of a Regulatory Body) where the Officer has reason to believe that the Officer will be required to appear before that inquiry or hearing relating to an act or omission of the Officer in providing services in their capacity as an officer of the Company or Relevant Company (as the case may be) during the Relevant Period; (c)conducting or preparing to conduct an action or proceeding which the Officer in good faith proposes to bring relating to an act or omission of the Officer in providing services in their capacity as an officer of the Company or Relevant Company (as the case may be) during the Relevant Period; or (d)any other purpose which the Company has provided written consent. |
Proceedings | (a)any investigation, hearing, inquiry or review undertaken by a court, arbitrator, mediator or tribunal, governmental, administrative or Regulatory Body, or public authority; and (b)any procedural step relating to such a hearing, conference, dispute, inquiry or investigation, under or in respect of which the Officer is being examined or is involved because the Officer is or was an officer of the Company or a Relevant Company (as the case may be) in the Relevant Period. |
Page | 5
Defined term | Meaning |
Protection Period | in relation to the Company and each Relevant Company (as the case may be) the period commencing on the Appointment Date and ending on the later of: (a)the date which is 7 years after the Officer ceases to hold office as an officer of the Company or the Relevant Company (as the case may be); and (b)the date any Proceedings commenced during the period specified in paragraph (a) have been finally resolved. |
Regulatory Body | an entity constituted under the laws of Australia or any other jurisdiction which has the power to regulate the conduct and affairs of a Group Entity and the Officer and shall include (without limitation) the Australian Securities and Investment Commission, the Australian Competition and Consumer Commission and the Australian Tax Office. |
Related Body Corporate | has the meaning given to it in section 50 of the Corporations Act, and includes a Subsidiary and an Affiliate. |
Relevant Company | each Related Body Corporate of the Company of which the Officer is considered an officer from time to time. |
Relevant Period | the period commencing on the Appointment Date and ending on the date the Officer ceases to act as an officer of the Company or the Relevant Company (as the case may be). |
Subsidiary | has the meaning given in section 9 of the Corporations Act and refers to any corporation which before, at or after the date of this deed was, is or becomes a Subsidiary of the Company. |
1.2Interpretation
In this deed, unless the context requires otherwise:
(a)terms used from the Corporations Act have the meaning given under section 9 of the
Corporations Act;
(b)words importing the singular include the plural and vice versa;
(c)words importing a gender include any gender;
(d)a reference to a thing (including, but not limited to, a right) includes any part of that
thing;
(e)a reference to a right includes a remedy, power, authority, discretion or benefit;
(f)other parts of speech and grammatical forms of a word or phrase defined in this deed
have a corresponding meaning;
(g)a reference to a clause or party is a reference to a clause of, and a party to, this deed;
Page | 6
(h)a reference to a document or deed includes all amendments or supplements to, or
replacements or novations of, that document or deed;
(i)a reference to the Officer includes the Officer’s personal representatives;
(j)a reference to a statute, legislation, regulation, proclamation, ordinance or by-law
includes all statutes, regulations, proclamations, ordinances or by-laws amending,
consolidating or replacing it, and a reference to a statute includes all regulations,
proclamations, ordinances and by-laws issued under that statute;
(k)a reference to a party to a document includes that party’s successors and permitted
assignees;
(l)a rule of construction does not apply to the disadvantage of a party because that
party was responsible for the preparation of this deed or any part of it; and
(m)if a day for payment upon which an obligation must be performed is not a Business
Day the payment is due on the next Business Day.
2Indemnity |
1.1General indemnity
(a)To the maximum extent permitted by law, the Company indemnifies the Officer on a
full indemnity basis, with effect from the Appointment Date and on the terms set out in
this deed, against:
(i)all Liabilities incurred by the Officer as an officer of the Company or of a
Related Body Corporate;
(ii)all Liabilities incurred by the Officer in relation to actual, threatened or
potential Proceedings; and
(iii)subject to clause 2.1(b), all Liabilities incurred by the Officer in the Officer’s
capacity as an officer of the Company or of a Related Body Corporate for
Proceedings brought by the Officer against third parties in order to protect the
Officer’s interests or reputation. This indemnity includes, without limitation, a
liability for reasonable legal costs on a solicitor and own-client basis.
(b)The Company will only indemnify the Officer under clause 2.1(a)(iii) if the Officer first
obtains the written consent of the Board which:
(i)must not be unreasonably withheld or delayed; and
(ii)may be provided subject to any conditions the Board considers appropriate
(acting reasonably) including, without limitation, as to the treatment of all or
any damages or other compensation received by the Officer in respect of any
Proceedings.
1.2Continuing indemnity
(a)The indemnity in clause 2.1 is an irrevocable, unconditional, continuing and principal
obligation of the Company despite:
(i)the resignation or removal of the Officer as an officer of the Company;
(ii)the settlement of any dispute between the Officer and the Company or a third
party;
(iii)any amendment or variation to, or replacement of, the Constitution;
(iv)any intermediate payments, settlement of accounts or payments;
(v)laches, acquiescence or delay on the part of the Officer;
(vi)the death, bankruptcy, insolvency or liquidation of any person or corporation;
or
Page | 7
(vii)the occurrence of any other thing, including any other thing which might
otherwise affect the indemnity whether at law or in equity,
and remains in full force and effect until released by the Officer.
(b)The Company shall not be obliged to indemnify the Officer under this deed where the
Officer fails to perform any of the obligations set out in clause 3.5 to the material
prejudice of the Company.
(c)The indemnity in clause 2.1 enures to the benefit of the Officer’s estate.
1.3Additional indemnity
The indemnity in clause 2.1 is in addition to any indemnity contained in the Constitution from
time to time.
3Conduct and obligations |
1.1Notification
The Officer must give notice to the Company as soon as reasonably practicable after the
Officer becomes aware of any facts, matters, circumstances, or any threatened or pending
Claim against the Officer or decision to make a Claim against a third party, which could give
rise to a Claim for indemnification under this deed.
1.2Advancement or payment of costs
(a)On the Officer’s request, the Company will advance to or pay on behalf of the Officer,
reasonable costs incurred or expected to be incurred by the Officer (whether legal or
otherwise) in connection with Proceedings on terms determined by the Board except
that any such advance will be interest free and on an unsecured basis.
(b)The Officer must furnish the Company with invoices or other relevant evidence
satisfactory to the Company of the costs incurred or expected to be incurred for the
purposes of clause 3.2(a).
(c)If the Company has advanced an amount for costs under clause 3.2(a), the amount of
the advance will be in part satisfaction of the Company’s obligation to indemnify the
Officer and will cease to be repayable unless it is subsequently found that the Officer
was not entitled to be indemnified for those costs.
(d)If the Officer has a right of recovery from a third party in respect of some or all of the
Liabilities, the Officer must use best efforts to exercise the right of recovery within 10
days of payment being received and account to the Company for any such amount.
(e)If the Company has advanced or otherwise paid an amount under clause 3.2(a) and it
is subsequently found that the Officer was not entitled to the advancement or
payment of those costs, the Officer must repay this amount to the Company within 30
days of a request from the Company being received by the Officer.
1.3Proceedings
(a)The Company may:
(i)assume the conduct, negotiation or defence of a Claim;
(ii)institute legal proceedings (including a counterclaim or cross claim) in relation
to a Claim; and
(iii)subject to clause 3.4, retain lawyers to act on behalf of both the Company
and the Officer in relation to the Claim,
(iv)and, when it does so, the conduct of the claim will be under the management
and control of the Company or its insurers (acting reasonably).
(b)The Company must:
Page | 8
(i)notify the Officer as soon as reasonably practicable if it intends to take any
action under clause 3.3(a);
(ii)consider the reputation of the Officer in acting under clause 3.3(a); and
(iii)not unreasonably withhold or delay its decision under clause 3.3(b)(ii).
(c)If the Company does not elect to take control of the conduct of proceedings under
clause 3.3(a), the Officer must ensure that the Company is kept fully informed of any
actual or proposed developments (including, without limitation, any meetings) and is
provided with copies of all material correspondence and documentation relating to
such third party Claim or action, and such other information, assistance and access to
records and personnel as the Company reasonably requires, other than any
correspondence, documents or information to which any legal professional privilege
attaches for the benefit solely of the Officer.
1.4Legal representation
(a)The Officer may obtain independent legal representation in respect of the conduct,
negotiation or defence of any advice, Claim or Proceeding against the Officer as a
result of or arising from being an officer of the Company or a Related Body Corporate.
(b)The Company will reimburse the Officer within 10 days of receipt of a written request
for reimbursement from the Officer for expenses payable by the Company under this
deed to the extent that those expenses are:
(i)reasonable and incurred prior to the Company assuming conduct of the
Claim;
(ii)incurred with the Company’s prior written authority (which must not be
unreasonably withheld or delayed); and
(iii)reasonable and incurred where it could be reasonably expected that a conflict
between the interests of the Officer and the Company (or a Related Body
Corporate) would arise should the same lawyers act on behalf of both parties.
1.5Officer’s obligations
To the extent permitted by law, the Officer must:
(a)give notice to the Company promptly on becoming aware of any claim, or any
circumstances which could reasonably be expected to give rise to any claim, that may
give rise to a right for the Officer to be indemnified under this deed;
(b)take any reasonable action to avoid, resist, dispute, bring an appeal in, compromise
or defend any Claim;
(c)not make an admission of liability or payment in respect of or settle or compromise
any Claim without the Company’s prior written consent (which must not be
unreasonably withheld or delayed);
(d)render all reasonable assistance to the Company in the conduct of any Claim,
including (but not limited to) providing documents, authorities and directions that the
Company requires to prosecute or advance any cross claim or counterclaim; and
(e)on request by the Company, do anything reasonably necessary or desirable to enable
the Company (so far as possible) to be subrogated to and enjoy the benefits of the
Officer’s rights in relation to any cross-claims or any Claims against any third party
and render any assistance that is reasonably requested by the Company for the
purpose,
and the Company is not obliged to indemnify the Officer under this deed or otherwise where
the Officer fails to perform any of these obligations to the material prejudice of the Company.
4Insurance |
Page | 9
1.1Obligation to maintain D&O Policy
(a)Subject to clause 4.2 and to the extent permitted by law, the Company must:
(i)at all times during the Protection Period maintain a D&O Policy consistent
with clause 4.2; and
(ii)not undertake any actions which will, or is likely to, reduce or invalidate the
cover under the D&O Policy, other than making a claim.
1.2Terms and conditions of Policy
(a)Subject to law, the terms of the D&O Policy must, to the extent that such a policy is
available from a reputable insurance company at reasonable commercial rates:
(i)cover the Liabilities which may be incurred by the Officer during the
Protection Period arising from the Officer acting as an Officer of the
Company;
(ii)provide for the terms, conditions, exclusions and additional cover (including
premiums, insuring clauses, exclusions and excess amounts) as are
reasonably appropriate for a company of similar industry and annual revenue;
and
(iii)be on terms which are not materially less favourable or at least as
comprehensive as the insurance policies provided to the then current
directors and officers of the Company from time to time during the Protection
Period.
1.3Obligations of Officer
(a)The Officer undertakes to comply with the obligations and requirements provided for
under the D&O Policy and to take reasonable steps to enable the Company to
maintain the D&O Policy under this deed.
(b)The Officer undertakes to disclose to the Company all known relevant matters which
may lead to a claim being made against the Company or the Officer, as soon as
practicable after becoming aware of such matters.
(c)To the extent required to comply with its disclosure obligations in relation to the D&O
Policy, the Company will seek information from the Officer within a reasonable period
before the renewal of the policy.
(d)The Officer acknowledges that negotiations undertaken by the Company on the terms
of the D&O Policy may result in the insurer varying the terms of the D&O Policy.
1.4Obligations of Company
(a)The Company must provide the Officer with a copy of the D&O Policy and certificate
of insurance upon request by the Officer.
(b)The Company must:
(i)to the maximum extent permitted by law, pay the cost of any premiums under
the D&O Policy; and
(ii)notify the Officer upon becoming aware that the D&O Policy has been
cancelled, not renewed, or there is a material reduction in the terms of the
D&O Policy.
5Taxation and costs |
1.1Taxation and duty
(a)If a government authority imposes any tax on a sum paid to the Officer under this
deed, then the Company or the Relevant Company (as the case may be) must
increase the amount paid to the Officer so that the net amount to which the Officer
becomes entitled after deduction of all applicable taxes is equal to that which would
have been payable under the deed had no such tax been imposed.
Page | 10
(b)The Company or the Relevant Company (as the case may be) must pay any stamp
duty chargeable on this deed.
1.2Costs
Each party must bear its own costs of negotiating, preparing and executing this deed.
6General |
1.1Governing law and jurisdiction
This deed is governed by the laws of Victoria, Australia and each party irrevocably submits to
the non-exclusive jurisdiction of the courts of Victoria, Australia.
1.2Counterparts
This deed may be executed in counterparts. All executed counterparts constitute one
document.
1.3Unenforceable provision
If a provision in this deed is wholly or partly invalid or unenforceable in any jurisdiction, the
provision or part of it that is invalid or unenforceable must, to that extent and in that
jurisdiction, be treated as deleted from this deed. Any provision removed under this clause 6.3
does not affect the validity or enforceability of the remaining provisions in that jurisdiction or
any other jurisdiction.
1.4Survival
Each obligation of confidence and indemnity in this deed is a continuing obligation, separate
and independent from the other obligations and survives termination of this deed for any
reason.
1.5Further action
Each party must, at its own expense, do all things reasonably necessary (including executing
all documents) to give full effect to this deed and the transactions contemplated by it.
1.6Exclusion of moratoria
Any statute, moratorium or other governmental order that prejudicially affects the rights,
powers or discretions of the parties under this deed does not apply to this deed unless the
application is mandatory.
1.7Variation and waiver
(a)This deed may only be varied in writing signed by each party.
(b)A party does not:
(i)waive a right, power, or remedy unless it does so in writing and any such
waiver is only effective for the specific instance for which it is given; or
(ii)waive a right, power, or remedy if it fails to exercise or delays in exercising
the right, power or remedy.
(c)A single or partial exercise of a right by a party does not prevent another further
exercise of that right, power, or remedy.
1.8Notices
Any notices or other communications under this deed must be in writing addressed to the
recipient as detailed on this deed and hand delivered or sent by prepaid post or facsimile to
the number as specified to the sender by notice.
Page | 11
1.9Entire Agreement
This deed embodies the entire agreement between the parties with respect to the subject
matter of this deed and supersedes any prior negotiation, arrangement, understanding or
agreement with respect to the subject matter of any term of this deed.
Execution Page |
EXECUTED AS A DEED
Executed by Telix Pharmaceuticals Limited ACN 616 620 369 in accordance with the Corporations Act 2001 (Cth) by being signed by the following officers: | |||
Director Christian Behrenbruch | Director/Secretary Genevieve Ryan | ||
\ | |||
Name (please print) | Name (please print) |
Signed, sealed and delivered by insert officer name in the presence of: | |||
Insert Officer name | Witness | ||
Name (please print) |
Exhibit 8.1
Subsidiaries of Telix Pharmaceuticals Limited
Name of Entity | State or Jurisdiction of Incorporation or Organization | Percentage Ownership and Voting Interest (%) | ||
Telix Pharmaceuticals Holdings Pty Ltd | Australia | 100 | ||
Telix Pharmaceuticals International Holdings Pty Ltd | Australia | 100 | ||
Telix Pharmaceuticals Australia Holdings Pty Ltd | Australia | 100 | ||
Telix Pharmaceuticals (Innovations) Pty Ltd | Australia | 100 | ||
Telix Pharmaceuticals (ANZ) Pty Ltd | Australia | 100 | ||
Telix Pharmaceuticals (Corporate) Pty Ltd | Australia | 100 | ||
Telix Pharmaceuticals (NZ) Limited | New Zealand | 100 | ||
Telix Pharma Japan KK | Japan | 100 | ||
Telix Pharmaceuticals (Singapore) Pte Ltd | Singapore | 100 | ||
Telix Pharmaceuticals (US) Inc. | Delaware | 100 | ||
Telix Optimal Tracers LLC | Delaware | 100 | ||
Telix Pharmaceuticals (Canada) Inc. | Canada | 100 | ||
Telix Innovations SA | Belgium | 100 | ||
Telix Pharmaceuticals (Germany) GmbH | Germany | 100 | ||
Telix Pharmaceuticals (Switzerland) GmbH | Switzerland | 100 | ||
Telix Pharmaceuticals (Belgium) SRL | Belgium | 100 | ||
Lightpoint Surgical Ltd | United Kingdom | 100 | ||
Lightpoint Surgical Spain S.L. | Spain | 100 | ||
Rhine Pharma GmbH | Germany | 100 | ||
Therapeia GmbH & Co. KG | Germany | 100 | ||
Therapeia Verwaltungs- GmbH | Germany | 100 | ||
Telix Pharmaceuticals (France) SAS | France | 100 | ||
Telix Pharmaceuticals (UK) Ltd | United Kingdom | 100 | ||
Telix IsoTherapeutics Group Inc. | Delaware | 100 | ||
Telix ARTMS Inc. | Canada | 100 | ||
ARTMS US, Inc. | Delaware | 100 | ||
Telix QSAM, Inc. | Delaware | 100 | ||
QSAM Therapeutics Inc. | Texas | 100 | ||
Telix Innovations RPH Participações Ltda. | Brazil | 51 | ||
RLS (USA) Inc. | Delaware | 100 | ||
Las Vegas Radiopharmacy, Inc. | Delaware | 100 | ||
Telix Targeting Technologies, Inc. | Delaware | 100 |
Exhibit 11.1

Securities Dealing Policy
Telix Pharmaceuticals Limited
Adopted by the Board
effective on 11 December 2025
2
Table of Contents
1.1Insider trading prohibition . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . | |
1.2Extra-territorial application . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . | |
1.3Front Page Test . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . | |
1.1No dealing during Blackout Periods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . | |
1.2Trading Windows . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . | |
1.3Connected Persons . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . | |
1.4No short-term or speculative dealing . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . | |
1.5No short selling . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . | |
1.6No hedging of Telix Securities . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . | |
1.7No margin lending . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . | |
schemes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . | |
1.2Other exemptions - exceptional circumstances . . . . . . . . . . . . . . . . . . . . . . . . | |
1.3Other exemptions - excluded dealings . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . | |
1.1Prior approval . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . | |
1.2Approval request conditions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . | |
1.3Duration of approval . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . | |
Directors) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
3
1 |
2Purpose and Objectives |
Telix Pharmaceuticals Limited (Telix) and its Employees and Partners are bound by applicable laws, regulations and
Listing Rules governing the conduct for buying, selling and otherwise dealing in Telix Securities. The purpose of this
Securities Dealing Policy (Policy) is to:
(a)explain the law and associated policy relating to insider trading, including types of conduct in dealing in Telix
Securities that are prohibited under the Corporations Act 2001 (Cth) (Corporations Act) and the U.S. Securities
Exchange Act of 1934, as amended, (U.S. Securities Exchange Act) and to whom such prohibitions apply; and
(b)establish a best practice procedure for dealing in Telix Securities that protects Telix and its worldwide affiliates or its
related bodies corporate (together, the Group) and its Employees and Partners in respect of dealing in Telix
Securities and other securities.
3Policy Application |
2.1 Scope
This Policy applies to the Group worldwide, its directors, officers, executive and senior management, and other
employees (Employees) as well as Telix’s consultants, contractors, vendors, collaboration partners or advisors, acting
on behalf of Telix or the Group with access to Inside Information (Partners).
2.2 Definitions
For the purposes of this Policy, the following definitions apply:
Board means the Telix Board of Directors.
Blackout Period has the meaning set out in section 4.1.
Connected Person is a person or entity that is connected to another person or entity in a way that allows for influence
over dealings, including but not limited to:
(a)individuals with close family ties, business partners, and associated companies or trusts;
(b)a company or any other entity which the Employee or Partner has an ability to control, including its directors and
officers.
An Employee or Partner must:
(a)inform their Connected Persons about this Policy; and
(b)communicate on behalf of their Connected Persons in relation to requests for approval (as applicable under this
Policy).
Director means a Director of Telix.
Employees has the meaning set out in section 2.1.
ESPP has the meaning set out in section 5.1.
Front Page Test has the meaning set out in section 3.3.
Group has the meaning set out in section 1.
Inside Information is information that:
(a)is not generally available to the market; and
(b)if it were generally available to the market, a reasonable person would expect it to have a material effect (upwards
or downwards) on the price or value of a security.
Information is regarded as being likely to have a material effect if it would, or would be likely to, influence persons who
commonly invest in securities or other traded financial products in deciding whether or not to deal in the security.
5
Some examples which may constitute Inside Information include:
(a)Telix’s financial performance;
(b)Telix considering a major acquisition or disposal of assets, or a takeover or merger;
(c)an undisclosed significant change in Telix’s market share;
(d)undisclosed material operational or regulatory developments;
(e)changes in the capital structure of Telix, including proposals to raise additional equity or increase debt;
(f)major new initiatives or proposed changes in the nature of the business of Telix;
(g)changes to the Board or executive key management personnel;
(h)likely entry into (or loss of) a material contract or government approval;
(i)likely receipt of grant (or decline) of a marketing authorization approval by a regulatory agency;
(j)a proposed dividend or other distribution or a change in dividend policy;
(k)actual or threatened major litigation or governmental proceedings or investigations, or the actual or potential
settlement of such litigation, proceedings or investigations;
(l)the existence of a special blackout period;
(m)significant cybersecurity incidents;
(n)other significant unexpected liabilities.
Listing Rules means the listing rules of the Australian Securities Exchange (ASX), the Nasdaq Stock Market, the
Singapore Exchange (as applicable) and any other exchange on which Telix Securities may from time to time be traded.
Partner has the meaning set out in section 2.1.
Restricted Persons for the purposes of this Policy are:
(a)each Director;
(b)the Group Company Secretary of Telix;
(c)each member of the Telix Group Executive Team and key management team members and their respective direct
reports; and
(d)any other Employee or Partner designated by the Group Company Secretary from time to time.
A list of Restricted Persons shall be maintained by People & Culture with oversight by the Group Company Secretary
Telix means Telix Pharmaceuticals Limited.
Telix Securities means shares, American Depositary Shares representing shares, and other instruments considered to
be a security for the purposes of the Corporations Act or Listing Rules. The definition of a security is complex and
includes not only ordinary shares but also options, performance share appreciation rights and rights over ordinary
shares, as well as exchange traded options and warrants.
Telix Share Plan Derivative has the meaning set out in section 5.1.
Trading Window means times other than the Blackout Periods in which Employees and Partners may deal in Telix
Securities (provided they are not otherwise in possession of Inside Information).
4Insider Trading - Law |
1.1Insider trading prohibition
Compliance with insider trading laws and this Securities Dealing Policy is each individual’s responsibility. The law
prohibits insider trading and imposes substantial penalties, including imprisonment, for breaching applicable laws. It is
therefore important for all Employees and Partners to understand what constitutes insider trading and the types of
conduct that are prohibited.
6
Employees and Partners must not deal in Telix Securities (or securities of another entity) at any time if the dealing would
constitute insider trading. This will typically occur where Employees or Partners are in possession of Inside Information.
This means that an Employee or Partner who possesses Inside Information in relation to Telix Securities or another
entity’s securities must not:
(a)apply for, acquire or dispose of those securities, or agree to do so (whether on their own behalf or on behalf of
another person);
(b)procure, encourage, incite or induce any other person (for example, a family member, friend, or advisor) or any
entity owned or controlled by, or subjected to influence by, an Employee or Partner to do any of the above things;
or
(c)directly or indirectly communicate the Inside Information to any other person, if the Employee or Partner knows or
ought reasonably to know that the other person may use the information to do any of the above things.
1.2Extra-territorial application
Under the Corporations Act, the prohibition against insider trading applies to acts within Australia and acts outside
Australia that involve the securities of companies that are Australian or do business in Australia.
The law against insider trading applies to conduct relating to dealing in Telix Securities which occurs outside Australia
and within Australia.
In Australia and the United States, insider trading can result in administrative, civil or criminal proceedings which can
result in significant fines and civil penalties, being barred from service as an officer or director of a company, or
imprisonment.
Employees and Partners must also comply with any applicable obligations or requirements under local law in their
jurisdiction (and any jurisdiction in which they may from time-to-time travel to, or be located in, in connection with Group
business).
1.3Front Page Test
It is important that public confidence in Telix is maintained. It would be damaging to Telix’s reputation if the market or the
general public perceived that Employees or Partners might be taking advantage of their position in the Group to make
financial gains (by dealing in securities on the basis of Inside Information).
As a guiding principle, Employees or Partners should ask themselves:
If the market was aware of all the current circumstances, could I be perceived to be taking advantage of my position in
an inappropriate way? How would it look if the transaction were reported on the front page of the newspaper? (Front
Page Test).
If the Employee or Partner is unsure, they should consult the Group Company Secretary or the Group General Counsel.
Where any approval is required for a dealing under this Policy under section 6.1, approval will not be granted where the
dealing would not satisfy the Front Page Test.
5Dealing in Telix Securities - Policy |
In addition to complying with the law in relation to insider trading (under section 3), Employees and Partners must adhere
to the following policy requirements.
1.1No dealing during Blackout Periods
There are certain periods in the year where Employees and Partners must not deal in Telix Securities due to the
proximity of those periods to the release of Telix’s financial or trading results - and therefore a heightened risk of actual or
perceived insider trading (Blackout Periods).
Blackout Periods are set out below. Even when a Blackout Period is not operating, Employees or Partners may be
restricted from dealing in Telix Securities by applicable insider trading laws.
7
Event | Blackout Period |
Release of Full Year Results | From the close of trading on the ASX on 31 December each year until the start of trading on the day following the release. |
Release of Half Year Results | From the close of trading on the ASX on 30 June each year until the start of trading on the day following the release. |
Any other special blackout period that the Board specifies from time to time. | |
Restricted Persons are also prohibited from dealing in Telix Securities during the following periods.
Event | Additional Blackout Period |
Release of Q1 Business Update | From the close of trading on the ASX on 31 March each year until the start of trading on the day following the release. |
Release of Q3 Business Update | From the close of trading on the ASX on 30 September each year until the start of trading on the day following the release. |
Any other special blackout period that the Board specifies from time to time for Restricted Persons. | |
1.2Trading Windows
Trading Windows are times other than the Blackout Periods described in section 4.1 above. Employees and Partners
may only deal in Telix Securities during Trading Windows when they are not in possession of Inside Information.
Directors (and their Connected Persons) and Employees and Partners wishing to deal in Telix Securities during a
Blackout Period or on a short-term basis in exceptional circumstances may only deal in Telix Securities in such
circumstances if prior approval is sought in accordance with the procedure set out in section 6.1.
1.3Connected Persons
Employees and Partners must take appropriate steps to ensure that their Connected Persons only deal in Telix
Securities in circumstances where the Employee or Partner to whom they are connected would be permitted to deal
under this Policy. For example, not trading during Blackout Periods unless in exceptional circumstances (section 5.2),
and approval is obtained in accordance with the procedure outlined in section 6.1.
1.4No short-term or speculative dealing
Employees and Partners must not buy and sell Telix Securities on a short-term basis (that is, within a three-month
period), except Telix Securities may be sold in exceptional circumstances and provided prior approval is sought and
granted in accordance with the procedure set out in section 6.1.
1.5No short selling
Employees and Partners must not engage in any short selling of Telix Securities. Short selling occurs when a person
sells financial products they do not own with a view to repurchasing them later at a lower price.
1.6No hedging of Telix Securities
Employees and Partners must not engage in hedging of Telix Securities. Hedging is a form of dealing and includes
entering into transactions in financial products that operate to limit the economic risk associated with holding Telix
Securities.
8
1.7No margin lending
Employees and Partners must not obtain margin loans using Telix Securities (either solely or as part of a portfolio) as
security for the loans, or enter into any other secured financing arrangements in respect of Telix Securities.
6Exemptions |
1.1Exemption for participation in Telix employee equity plans or similar schemes
The restriction in section 4.1 does not apply to:
(a)participation in a Telix employee equity incentive plan;
(b)the exercise of any option, warrant, right or any other class of convertible security issued under a Telix employee
equity plan (Telix Share Plan Derivative); provided, however, that any such exercise and subsequent conversion
or issue of Telix Securities is not at a time when the Employee is in possession of Inside Information in
circumstances where Telix is required to dispose of Telix Securities on behalf of the Employee to satisfy regulatory
obligations or this action would otherwise be in breach of this Policy, including section 4.1. Subsequent dealings
with Telix Securities issued on exercise or conversion of any Telix Share Plan Derivative are always subject to this
Policy, including section 4.1; and
(c)purchases of ordinary shares or American Depositary Shares through periodic, automatic payroll contributions to a
Company employee share or stock purchase plan (ESPP). However, electing to enroll in an ESPP, making any
changes in elections under an ESPP or terminating contributions under an ESPP are not permitted during a
Blackout Period or while the Employee is in possession of Inside Information. In addition, any ordinary shares or
American Depositary Shares issued under an ESPP are subject to this Policy.
1.2Other exemptions - exceptional circumstances
In exceptional circumstances, Employees and Partners may be given prior written approval to dispose of (but not
acquire) Telix Securities where they are not in possession of Inside Information and they would otherwise be restricted by
this Policy due to the application of a Blackout Period (section 4.1) or the general prohibition on short-term dealing
(section 4.4).
Exceptional circumstances may include severe financial hardship, a requirement under a court order, court enforceable
undertaking or other legal or regulatory requirement (for example, a family law settlement).
All requests for approval must be sent to the Group Company Secretary in accordance with the procedure set out in
section 6.1.
1.3Other exemptions - excluded dealings
Without affecting the obligation of Employees, Partners and/or their Connected Persons to ensure they comply at all
times with insider trading laws, the Policy does not apply to the following categories of trades:
(a)acquisition of Telix Securities through a dividend reinvestment plan;
(b)acquisition of Telix Securities through a share purchase plan available to all shareholders;
(c)acquisition of Telix Securities through a rights issue or other pro rata entitlement offer available to all shareholders;
(d)disposal of Telix Securities through the acceptance of a takeover offer, scheme of arrangement or equal access
buy-back;
(e)dealings that result in no effective change to the beneficial interest in Telix Securities; and
(f)trading under a pre-approved non-discretionary trading plan, where the Employee did not enter into the plan or
amend the plan during a Blackout Period, the plan does not permit the Employee to exercise any influence or discretion
in relation to trading under the plan, and the plan cannot be cancelled during a Blackout Period or while the Employee is
in possession of Inside Information. Telix may require additional terms or restrictions for pre-approved non-discretionary
trading plans in compliance with applicable laws, including the Corporations Act and the U.S. Securities Exchange Act.
1 With each trade request, Directors should include as many details as possible in order to give the relevant approver full
context for the trade, including:
(a)number of securities to be acquired or disposed;
(b)timing for the trade;
(c)whether the securities are held directly or indirectly through one or more Connected Persons;
(d)the names of the Connected Persons and relationship to the Director;
(e)confirmation that the Director is not in possession of any Inside Information in relation to the securities; and
(f) such other information as may be requested or required by the relevant approver in connection with the request.
If approval is granted, as soon as possible following confirmation of trade, the Director must send the Group Company
Secretary confirmation in writing, including a copy of the trade statement or other documentation provided by the broker
(as applicable). Prompt notification to the Group Company Secretary is essential to assist Telix in complying with its
disclosure obligations under the Corporations Act, the U.S. Securities Exchange Act, the Listing Rules, the U.S. federal
securities laws and related compliance policies.
2 Approval can be sought and granted only when neither the Director nor Telix holds Inside Information or are otherwise
restricted under this policy, including section 4.1. Any on-market purchase by Telix to satisfy its obligation to provide Telix
Securities under the Non-Executive Director Rights Plans will only occur in those same circumstances.
3 Approval to dispose of Telix Securities during a Blackout Period or on a short-term basis in exceptional circumstances
will only be considered if the written request to the relevant approver is accompanied by:
(a)details of the proposed dealing including the number of Telix Securities to be disposed and date for executing the
proposed dealing;
(b)confirmation that the Employee or Partner is not in possession of Inside Information in relation to Telix Securities;
and
(c)sufficient evidence (in the opinion of the person providing clearance) that the disposal is the most reasonable
course of action available in the circumstances.
9
7Dealing in Telix Securities – Procedure |
1.1Prior approval
Prior approval to deal in Telix Securities:
(a)by Directors (including the CEO) and their Connected Persons during a Trading Window1;
(b)by Directors where Telix will purchase Telix Securities on-market to satisfy its obligations to provide such securities
to Directors under Telix’s Non-Executive Director Rights Plans2; or
(c)during a Blackout Period or on a short-term basis in exceptional circumstances, to the extent legally permitted at
the time of the request3, must be sought by the individual submitting a written request (including email) to deal in
Telix Securities to the relevant approver indicated below:
Person requiring approval | Approver | Notify |
Chair and their Connected Persons | Board or Chair of Audit and Risk Committee | Group Company Secretary |
Directors (including CEO) and their Connected Persons | Chair of Board | Group Company Secretary |
Company Secretary and their Connected Persons | CEO and Chair of Board | |
All other Employees and Partners | Group Company Secretary |
1.2Approval request conditions
10
Any request for approval to trade may be granted or refused without explanation.
If a request for approval to trade is granted, it may be withdrawn at any time prior to the order for dealing being lodged or
otherwise authorised, if new information comes to light or there is a change in the circumstances of Telix Securities.
If a request for approval to trade is refused, the decision is final and binding and the Employee or Partner who has
sought the approval must keep the information (of the refusal) confidential and not disclose it to anyone (other than to
their relevant Connected Person to which it relates, as applicable).
Approval is not an endorsement of any proposed dealing. All Employees and Partners are responsible for their own
investment decisions and compliance with the law (including the insider trading prohibition).
1.3Duration of approval
If approval to deal in Telix Securities is granted, the dealing must be conducted within three business days of the
approval. If the Employee or Partner does not deal in Telix Securities within this time period, the approval is no longer
effective and approval must be sought again.
1.4Prior notification and confirmation of trade – Restricted Persons (other than Directors)
Restricted Persons (other than Directors, including the CEO who must obtain prior approval under section 6.1) must
notify the Group Company Secretary both prior to the commencement of any trade, and as soon as practicable after
confirmation of any order to trade, including in respect of any of their Connected Persons. This is to assist Telix to comply
with its disclosure obligations under the Corporations Act, the U.S Securities Exchange Act, the Listing Rules, the U.S.
federal securities laws and related compliance policies.
8Non-compliance consequences |
It is each Employee or Partner’s responsibility (not Telix’s) to ensure that they and, where applicable, their Connected
Persons do not do any of the things prohibited by insider trading laws.
The consequences for breach of these laws can include both civil and criminal penalties, including imprisonment. This
prohibition against insider trading applies to all Employees and Partners at all times, including outside of Blackout
Periods and other ad-hoc restriction periods, and overrides all other provisions of this Policy, including any consent or
approval to trade which may be granted under this Policy.
It is important to note that international securities regulators, for example the U.S. Securities Exchange Commission
(SEC) and the European Securities and Markets Authority (ESMA) have authority in their own jurisdictions to commence
investigations and/or proceedings relating to insider trading by residents living in their jurisdiction.
Breaches of this Policy will be regarded by Telix as serious misconduct. In addition to the consequences applicable
under law, Employees or Partners who fail to adhere to the requirements of this policy may face disciplinary action,
including suspension or termination of employment, forfeiture of securities issued under any equity incentive plan of the
Group, and/or exclusion from participating in any equity incentive plan of the Group.
9Awareness and training |
The highest standards of corporate conduct are critical to the reputation of Telix. The Group Company Secretary is
responsible to ensure appropriate training and processes are in place across the Group to promote awareness and
compliance with this Policy. A copy of this Policy will be available on Telix’s website. It will be distributed to all Telix
Employees as part of employment induction and at regular intervals thereafter.
10Who should I contact? |
Any person who becomes aware of an actual or potential breach of this Policy should immediately report it to the Group
Company Secretary or the Group General Counsel.
Employees should also contact the Group Company Secretary if they are unsure about whether it is acceptable to deal
or communicate with others in relation to Telix Securities or other securities or if they have any other queries about this
Policy.
11
Telix encourages its Employees and Partners to seek their own professional advice before dealing in Telix Securities.
11Review |
The Board will review and update this Policy as required but at a minimum on an annual basis.
12Recent Change Summary |
Effective Date | Summary of Change | Author | Approval |
31 August 2017 | New Policy | Company Secretary | Approved by the Board |
11 April 2022 | Updated for changes in law and business since last update | Company Secretary | Approved by the Board |
29 May 2023 | Updated to clarify extension of application of policy to Connected Persons and changes in business since last update | Group Company Secretary | Approved by the Board |
22 August 2024 | Updated to current blackout periods following ASX relief from quarterly reporting in accordance with Listing Rules 4.7B and 4.7C | Group Company Secretary | Approved by the Board |
13 November 2024 | Updated to incorporate Nasdaq and SEC requirements following Telix’s listing on Nasdaq | Group Company Secretary | Approved by the Board |
12 December 2024 | Updated to include required references to and requirements of the employee share purchase plan | Group Company Secretary | Approved by the Board |
11 December 2025 | Updated to focus on principles and changes in governance practices | Group Company Secretary | Approved by the Board |
Exhibit 12.1
CERTIFICATION PURSUANT TO
RULES 13a-14(a) AND 15d-14(a) UNDER THE SECURITIES EXCHANGE ACT OF 1934,
AS ADOPTED PURSUANT TO SECTION 302 OF THE SARBANES-OXLEY ACT OF 2002
I, Christian Behrenbruch, certify that:
1.I have reviewed this annual report on Form 20-F of Telix Pharmaceuticals Limited;
2.Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state
a material fact necessary to make the statements made, in light of the circumstances under which such
statements were made, not misleading with respect to the period covered by this report;
3.Based on my knowledge, the financial statements, and other financial information included in this report,
fairly present in all material respects the financial condition, results of operations and cash flows of the
company as of, and for, the periods presented in this report;
4.The company’s other certifying officer(s) and I are responsible for establishing and maintaining disclosure
controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control
over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the company and
have:
(a)Designed such disclosure controls and procedures, or caused such disclosure controls and
procedures to be designed under our supervision, to ensure that material information relating to the
company, including its consolidated subsidiaries, is made known to us by others within those
entities, particularly during the period in which this report is being prepared;
(b)Designed such internal control over financial reporting, or caused such internal control over
financial reporting to be designed under our supervision, to provide reasonable assurance
regarding the reliability of financial reporting and the preparation of financial statements for
external purposes in accordance with generally accepted accounting principles;
(c)Evaluated the effectiveness of the company’s disclosure controls and procedures and presented in
this report our conclusions about the effectiveness of the disclosure controls and procedures, as of
the end of the period covered by this report based on such evaluation; and
(d)Disclosed in this report any change in the company’s internal control over financial reporting that
occurred during the period covered by the annual report that has materially affected, or is
reasonably likely to materially affect, the company’s internal control over financial reporting; and
5.The company’s other certifying officer(s) and I have disclosed, based on our most recent evaluation of
internal control over financial reporting, to the company’s auditors and the audit committee of the
company’s board of directors (or persons performing the equivalent functions):
(a)All significant deficiencies and material weaknesses in the design or operation of internal control
over financial reporting which are reasonably likely to adversely affect the company’s ability to
record, process, summarize and report financial information; and
(b)Any fraud, whether or not material, that involves management or other employees who have a
significant role in the company’s internal control over financial reporting.
Date: February 20, 2026 | By: | /s/ Dr. Christian Behrenbruch |
Dr. Christian Behrenbruch | ||
Managing Director & Group Chief Executive Officer | ||
(Principal Executive Officer) |
Exhibit 12.2
CERTIFICATION PURSUANT TO
RULES 13a-14(a) AND 15d-14(a) UNDER THE SECURITIES EXCHANGE ACT OF 1934,
AS ADOPTED PURSUANT TO SECTION 302 OF THE SARBANES-OXLEY ACT OF 2002
I, Darren Smith, certify that:
1.I have reviewed this annual report on Form 20-F of Telix Pharmaceuticals Limited;
2.Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state
a material fact necessary to make the statements made, in light of the circumstances under which such
statements were made, not misleading with respect to the period covered by this report;
3.Based on my knowledge, the financial statements, and other financial information included in this report,
fairly present in all material respects the financial condition, results of operations and cash flows of the
company as of, and for, the periods presented in this report;
4.The company’s other certifying officer(s) and I are responsible for establishing and maintaining disclosure
controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control
over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the company and
have:
(a)Designed such disclosure controls and procedures, or caused such disclosure controls and
procedures to be designed under our supervision, to ensure that material information relating to the
company, including its consolidated subsidiaries, is made known to us by others within those
entities, particularly during the period in which this report is being prepared;
(b)Designed such internal control over financial reporting, or caused such internal control over
financial reporting to be designed under our supervision, to provide reasonable assurance
regarding the reliability of financial reporting and the preparation of financial statements for
external purposes in accordance with generally accepted accounting principles;
(c)Evaluated the effectiveness of the company’s disclosure controls and procedures and presented in
this report our conclusions about the effectiveness of the disclosure controls and procedures, as of
the end of the period covered by this report based on such evaluation; and
(d)Disclosed in this report any change in the company’s internal control over financial reporting that
occurred during the period covered by the annual report that has materially affected, or is
reasonably likely to materially affect, the company’s internal control over financial reporting; and
5.The company’s other certifying officer(s) and I have disclosed, based on our most recent evaluation of
internal control over financial reporting, to the company’s auditors and the audit committee of the
company’s board of directors (or persons performing the equivalent functions):
(a)All significant deficiencies and material weaknesses in the design or operation of internal control
over financial reporting which are reasonably likely to adversely affect the company’s ability to
record, process, summarize and report financial information; and
(b)Any fraud, whether or not material, that involves management or other employees who have a
significant role in the company’s internal control over financial reporting.
Date: February 20, 2026 | By: | /s/ Darren Smith |
Darren Smith | ||
Group Chief Financial Officer (Principal Financial & Accounting Officer) |
Exhibit 13.1
CERTIFICATION PURSUANT TO
18 U.S.C. SECTION 1350, AS ADOPTED PURSUANT TO
SECTION 906 OF THE SARBANES-OXLEY ACT OF 2002
In connection with the annual report of Telix Pharmaceuticals Limited (the “Company”) on Form 20-F for the year
ended December 31, 2025 as filed with the Securities and Exchange Commission (the “Report”), I, Dr. Christian
Behrenbruch, Managing Director & Group Chief Executive Officer of the Company, certify pursuant to 18 U.S.C.
§1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, that, based on my knowledge:
1. | The Report fully complies with the requirements of Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended; and |
2. | The information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company. |
Date: February 20, 2026 | By: | /s/ Dr. Christian Behrenbruch |
Dr. Christian Behrenbruch | ||
Managing Director & Group Chief Executive Officer (Principal Executive Officer) |
Exhibit 13.2
CERTIFICATION PURSUANT TO
18 U.S.C. SECTION 1350, AS ADOPTED PURSUANT TO
SECTION 906 OF THE SARBANES-OXLEY ACT OF 2002
In connection with the annual report of Telix Pharmaceuticals Limited (the “Company”) on Form 20-F for the year
ended December 31, 2025 as filed with the Securities and Exchange Commission (the “Report”), I, Darren Smith,
Group Chief Financial Officer of the Company, certify pursuant to 18 U.S.C. §1350, as adopted pursuant to Section
906 of the Sarbanes-Oxley Act of 2002, that, based on my knowledge:
1. | The Report fully complies with the requirements of Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended; and |
2. | The information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company. |
Date: February 20, 2026 | By: | /s/ Darren Smith |
Darren Smith | ||
Group Chief Financial Officer (Principal Financial & Accounting Officer) |
Exhibit 15.1
CONSENT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
We hereby consent to the incorporation by reference in the Registration Statement on Form S-8 (No. 333-283917) of Telix Pharmaceuticals Limited of our report dated February 20, 2026 relating to the financial statements and the effectiveness of internal control over financial reporting, which appears in this Form 20-F.
/s/ PricewaterhouseCoopers
Melbourne, Australia
February 20, 2026
Melbourne, Australia
February 20, 2026
1
This page is required for Australian Disclosure Requirements and has been intentionally left blank
This page is required for Australian Disclosure Requirements and has been intentionally left blank
1

Telix Pharmaceuticals Limited
ACN 616 620 369
55 Flemington Road
North Melbourne,
Victoria, 3051
Australia
ASX ANNOUNCEMENT
Results Announcement for the Year Ended 31 December 2025
Melbourne (Australia) and Indianapolis, IN (U.S.A.) — 20 February 2026. Telix Pharmaceuticals Limited (ASX: TLX,
Nasdaq: TLX, Telix, the Company) announces its financial results for the year ended 31 December 2025.
As approved by the Board of Telix, and in accordance with ASX Listing Rule 4.3A, please find attached the
following for immediate release to the market:
•Appendix 4E; and
•Annual Report.
Annual General Meeting
Telix's Annual General Meeting (AGM) will be held on Thursday, 21 May 2026 at 10am (Melbourne time). Full
details of the agenda and instructions to participate in the AGM will be provided to shareholders when the Notice
of Meeting is released.
In accordance with Telix's Constitution, the closing date for Director candidate nominations is Thursday, 9 April
2026, being at least 30 business days before the AGM.
Authorized for lodgement by:

Genevieve Ryan
Company Secretary
2
Telix Pharmaceuticals Limited
ACN 616 620 369
55 Flemington Road
North Melbourne,
Victoria, 3051
Australia
Appendix 4E
Financial year ended
31 December 2025 |
Results announcement to the market
Current Reporting Period: | year ended 31 December 2025 |
Previous Reporting Period: | year ended 31 December 2024 |
This page and the following pages comprise the year end information given to the ASX under Listing Rule 4.3A.
The results are prepared in accordance with International Financial Reporting Standards (IFRS), and also comply
with Australian Accounting Standards. Amounts are presented in US dollars (US$).
Revenue and net profit/(loss)
2025 Result | Change | Change | Change | 2024 Result | |
US$'000 | US$'000 | % | US$'000 | ||
Revenue from contracts with customers | 803,794 | Up | 287,243 | 56 | 516,551 |
(Loss)/profit after income tax for the year attributable to members | (7,125) | Down | (40,810) | (121) | 33,685 |
Total comprehensive (loss)/income for the year attributable to members | (14,124) | Down | (50,090) | (139) | 35,966 |
Dividends
No dividend was proposed or paid. Should any dividends be paid in the future, no assurances can be given as to
the level of franking credits attaching to such dividends.
2025 | 2024 | |
Cents | Cents | |
(Loss)/profit per share | (2.11) | 10.17 |
Net tangible assets per share | (100.00) | 16.00 |
Dividend per share | - | - |
1 Includes $14.1 million of inventory for TLX250-Px (Zircaix®) commercial launch expensed to R&D. This expense arises
from commercial inventory produced in anticipation of Zircaix approval and will be reversed upon FDA approval if
received.
3
Brief explanation of results
Telix recorded an operating profit for the year of $29,776,000 (2024: profit of $55,177,000), primarily driven by
continued strong growth in U.S. sales of Illuccix and the launch of Gozellix. The Company generated total
revenue of $803,794,000 (2024: $516,551,000). Operating expenditure in the year totalled $408,397,000
(2024: $285,871,000). Included within operating expenditure was $171,249,0001 (2024: $127,930,000) related
to R&D activities for the Company's assets and development programs. Income tax expense for the year was
$1,859,000 (2024: $4,230,000).
For further commentary on the Company's results and other information required by Listing Rule 4.3A, please
refer to the Company's investor releases and Annual Report, including the Operating and financial review and
Financial report lodged with the ASX today.
This Appendix 4E should be read in conjunction with the Annual Report, including on Form 20-F, which includes:
- Item 18 Financial statements; and
- Other sections, comprising the Directors' report, as tabled below.
Australian Disclosure Requirements | Form 20-F Reference |
Principal activities | Item 4.B Business Overview See subheading "Principal Activities of the Company in the year under review" |
Review of operations and activities | Item 4.B Business Overview Item 5.A Operating Results |
Business strategies and prospects for future years | Item 4.B Business Overview Item 5.A Operating Results |
Business risks | Item 3.D Risk Factors |
Significant changes in the state of affairs | Item 5.E Critical Accounting Estimates See subheading - "State of Affairs" |
Events subsequent to the end of the financial year | Item 5.E Critical Accounting Estimates See subheading - "Events subsequent to the end of the financial year" |
Review of operations, likely developments and expected results; Business strategies and prospects for future years | Item 4.B Business Overview See subheadings - "Review of operations, likely developments and expected results; Business Strategies and prospects for future years" |
Environmental regulation and compliance | Item 4.B Business Overview See subheadings - "Environmental regulation and compliance; Sustainability report" |
Dividends | Item 8.A Consolidated Statements and Other Financial Information See subheading - "Dividends" |
Information on directors | Item 6.A Directors and Senior Management See subheading - "Directors and Key Management Personnel" Item 6.C Board Practices See subheadings - "Meetings and attendance" "Directors' interests in the securities of Telix"; and "Shares issued for acquisitions, on exercise of rights or options and lapse of options" |
Remuneration report | The Remuneration report starts at Item 6 and ends part way through Item 6.B as indicated |
Indemnification of officers | Item 6.C Board Practices See subheading - "Indemnification of officers" |
Indemnification of auditors | Item 6.C Board Practices See subheading - "Indemnification of auditors" |
4
Australian Disclosure Requirements | Form 20-F Reference |
Rounding | Item 6.C Board Practices See subheading - "Rounding" |
Corporate governance | Item 6.C Board Practices See subheading - "Corporate governance" |
Non-audit Services | Item 6.C Board Practices See subheading "Auditor independence and non-audit services" |
Auditor's independence declaration | Exhibit 99.2 |
Directors' Resolution | Item 6.B Compensation See subheading - "Directors' resolution" |
Audit report
This Appendix 4E is based on the audited Financial report for the year ended 31 December 2025, contained in
the attached Annual Report.
The Appendix 4E and Annual Report have been approved for release by the Board of Directors.

Genevieve Ryan
Company Secretary
20 February 2026